Генотип — все статьи и новости
Генотип — совокупность генов организма и их отношений между собой, в том числе аллели и сцепление генов в хромосоме. В самом узком смысле это понятие может подразумевать все аллели гена или группы генов, контролирующих какой-либо признак организма, а в более широком — совокупность всех наследственных признаков организма, записанных в его ДНК (включая некодирующие последовательности), которые определяют его фенотип — внешний вид и внутреннее строение.
Сам термин «генотип» вместе с терминами «ген» и «фенотип» ввел немецкий генетик Вильгельм Людвиг Иогансен в 1909 году в своей работе «Элементы точного учения наследственности». Генотип не следует путать с геномом: в первом случае речь идет о наследственной информации конкретного организма, во втором — о наборе признаков, свойственном целому виду.
На ранних этапах изучения генотипа считалось, что все гены в нем действуют отдельно и каждый из них соответствует одному белку, выполняющему определенную функцию в организме и проявляющемуся как конкретный признак, который может быть качественным или количественным (например, рост, умение E. coli расщеплять какое-то питательное вещество, цвет лепестков ночной красавицы или гладкость или морщинистость горошины в опытах Менделя). Еще в первой половине XX века этот принцип — «один ген — один фермент» — считался передовым. Благодаря ему у ученых появилась сама возможность заняться расшифровкой генетического кода. Однако в дальнейшем было доказано, что генотип — это единая система элементов, которые взаимодействуют на разных уровнях. Например, один ген может кодировать более одного белка, не вся ДНК состоит из кодирующих последовательностей (экзонов), не все некодирующие последовательности бесполезны. Тот же рост контролируется вовсе не одним геном. Кроме того, гены влияют друг на друга, в результате чего несколько генотипов могут давать один и тот же фенотип или же действие нескольких аллелей или разных генов суммируется и дает один признак. Среди этих механизмов: неполное доминирование, как у ночной красавицы, когда при скрещивании пурпурного и белого цветка мы получаем «среднее арифметическое» — розовые цветки у потомства; эпистаз, когда один ген подавляет другой; кодоминирование, определяющее наследование агглютининов и агглютиногенов, на основании которых впервые было выделено четыре группы крови человека.
coli расщеплять какое-то питательное вещество, цвет лепестков ночной красавицы или гладкость или морщинистость горошины в опытах Менделя). Еще в первой половине XX века этот принцип — «один ген — один фермент» — считался передовым. Благодаря ему у ученых появилась сама возможность заняться расшифровкой генетического кода. Однако в дальнейшем было доказано, что генотип — это единая система элементов, которые взаимодействуют на разных уровнях. Например, один ген может кодировать более одного белка, не вся ДНК состоит из кодирующих последовательностей (экзонов), не все некодирующие последовательности бесполезны. Тот же рост контролируется вовсе не одним геном. Кроме того, гены влияют друг на друга, в результате чего несколько генотипов могут давать один и тот же фенотип или же действие нескольких аллелей или разных генов суммируется и дает один признак. Среди этих механизмов: неполное доминирование, как у ночной красавицы, когда при скрещивании пурпурного и белого цветка мы получаем «среднее арифметическое» — розовые цветки у потомства; эпистаз, когда один ген подавляет другой; кодоминирование, определяющее наследование агглютининов и агглютиногенов, на основании которых впервые было выделено четыре группы крови человека.
Также у особей с одинаковым генотипом в ходе индивидуального развития организма могут проявиться разные фенотипы, так как некоторые гены проявляются только в определенных условиях. Кроме того, если у двух организмов одинаковый фенотип, это не значит, что у них идентичные генотипы (при полном доминировании мы получаем доминантный фенотип и у гетерозигот, и у доминантных гомозигот). При этом фенотип может меняться на протяжении жизни организма, а генотип в целом остается неизменным (не считая ошибок при копировании ДНК и других мутаций).
Изображение: OpenClipart-Vectors/Pixabay
Описание генов — Институт общей генетики
Приведенные данные о влиянии генов на особенности поведения получены при исследованиях больших групп добровольцев в разных странах. Гены влияют на психологические особенности человека, но не определяют их. Каждый из генов даёт очень небольшой вклад (обычно 2%-3%) в проявление тех или иных особенностей. Проявление генов может меняться в зависимости от условий среды.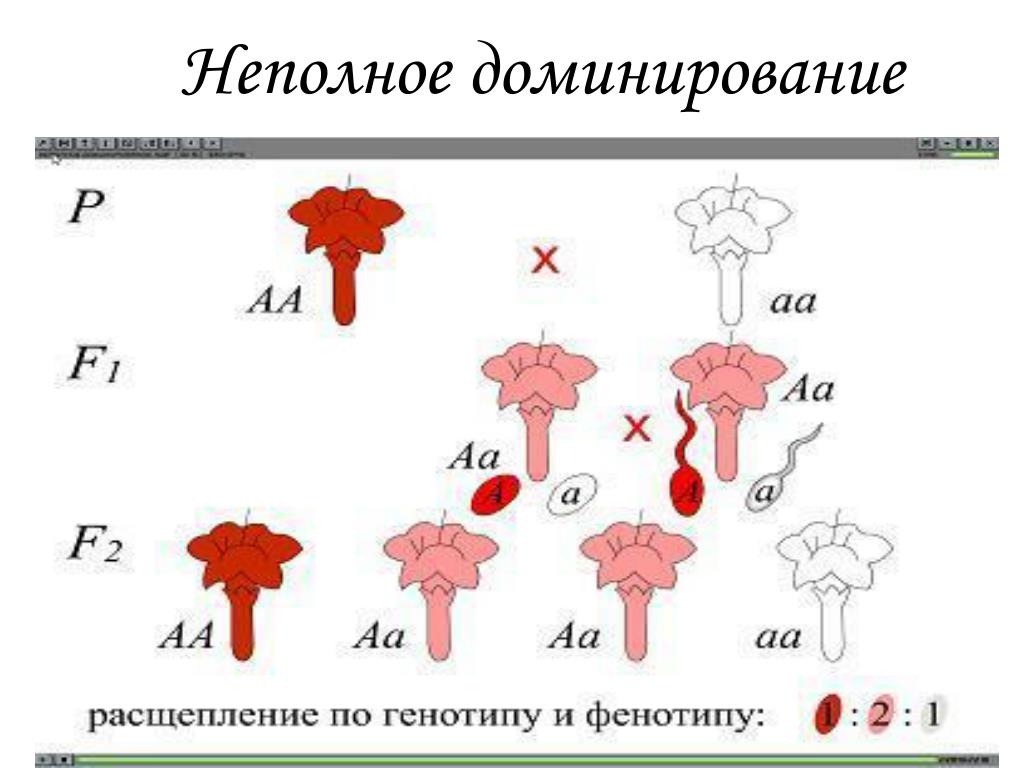 Например, один и тот же вариант гена может повышать вероятность развития депрессии в неблагоприятных условиях (при высоком уровне стресса) и снижать в благоприятных условиях. Такой вариант гена нельзя считать «плохим». Он обеспечивает более сильный ответ на условия среды. Носители другого варианта гена окажутся более стабильными по своим реакциям вне зависимости от изменений среды.
Например, один и тот же вариант гена может повышать вероятность развития депрессии в неблагоприятных условиях (при высоком уровне стресса) и снижать в благоприятных условиях. Такой вариант гена нельзя считать «плохим». Он обеспечивает более сильный ответ на условия среды. Носители другого варианта гена окажутся более стабильными по своим реакциям вне зависимости от изменений среды.
Схема передачи сигналов между нейронами (рис.1)
Более подробно о передаче сигнала между нейронами и структуре синапсов можно прочесть здесь
Более подробно о влиянии генов на поведение здесь.
Сокращенное латинское обозначение исследуемого варианта гена: 5’HTTLPR
Полное название гена: Транспортер серотонина
Функции: Регулирует передачу нервного импульса. Осуществляет обратный захват (перенос нейромедиатора серотонина, не связавшегося с рецепторами в синаптической щели, обратно в испустивший этот серотонин нейрон, чтобы зря не пропадал).
Синтезируется преимущественно в мозге и тонком кишечнике (там тоже есть серотонин, но выполняет другие функции).
Исследованные варианты гена: «короткий» S и «длинный» L варианты регуляторного участка гена. «Короткий» (S) менее активно работает, из-за этого синтезируется меньше белка-транспортера.
Возможные сочетания у индивида (генотипы): SS, SL, LL.
Особенности генотипов: S/S связан с большей вероятностью развития эмоциональных нарушений в неблагоприятной среде, например, депрессии (рис. 2) или посттравматического стрессового расстройства (ПТСР) при перенесенных стрессах (рис. 3).
Сокращенное латинское обозначение исследуемого варианта гена: COMT Val-158-Met
Полное название гена: Катехол-О-метилтрансфераза
Функции: Регулирует передачу нервного импульса, влияет на особенности эмоциональных реакций, участвует в метаболизме эстрогенов.
Синтезируется в мозге, печени, легких и других тканях.
Исследованные варианты гена: Val-158-Met (замена аминокислоты валина в позиции 158 на метионин), обозначемые латинскими буквами G и A (по названиям нуклеотидов гуанин и аденин в соответствующих этим аминокислотам позициях ДНК ).
Возможные сочетания у индивида (генотипы): AA, AG, GG.
Особенности генотипов: AA имеет более низкую активность по сравнению с генотипом GG медленнее разрушает катехоламины. Вариант AG имеет промежуточную активность. Люди с генотипом AA в среднем более позитивно реагируют на приятные события (возможно, потому, что медленнее разрушается дофамин). Генотип GG может быть связан с менее интенсивными переживаниями при негативных событиях, т.е. у обладателей этого генотипа удовлетворенность (или неудовлетворенность) жизнью может меньше зависеть от жизненных событий. Необходимо еще раз подчеркнуть, что вклад этих генов в вариации эмоционального состояния очень небольшой и зависит от взаимодействия с окружающими условиями.
Необходимо еще раз подчеркнуть, что вклад этих генов в вариации эмоционального состояния очень небольшой и зависит от взаимодействия с окружающими условиями.
Ученые объяснили, почему все клетки разные и почему человек не похож на мышь
Ученые впервые выяснили, как работают одни и те же гены в разных клетках человека и мыши. Российский участник проекта Fantom биоинформатик Всеволод Макеев рассказал «Газете.Ru» о задачах работы и о том, как она велась.
Основная загадка жизни
Прочтение последовательности букв в геноме человека еще не дает понимания того, как работает геном. Это не расшифровка генома, а, наоборот, зашифрованный текст, смысл которого мы пока не понимаем. Основная интрига заключается в том, что все клетки организма имеют одну и ту же ДНК, в которой содержится информация о кодировании определенных белков. Но клетки разных тканей разные, мышечные клетки не похожи на нервные или на клетки крови. В процессе развития каждый организм проходит путь от оплодотворенной яйцеклетки до взрослой особи и при этом все время меняется, а геном — нет.
В том, как это все регулируется, — в этом и состоит основная загадка жизни.
В последнее десятилетие усилия ученых были направлены на то, чтобы последовательно приближаться к пониманию того, как работает геном. О большом шаге в этом направлении рассказано в двух статьях последнем выпуске журнала Nature, где опубликованы результаты исследований в рамках программы Fantom. Название этой программы расшифровывается как Functional ANnoTation Of Mammals — функциональная характеристика генома млекопитающих.
Принципы работы генома универсальны что для мыши, что для человека — именно эти представители млекопитающих стали основными объектами исследований. У человека и мыши примерно один и тот же набор генов, в то же время мы совсем не похожи на мышь. Все дело в том, что гены человека регулируются по-другому, чем гены мыши. Ученые убедились в этом, нанеся на карту эти регуляторы и сравнив, как они работают у одного и другого вида.
«Исследования дают нам взгляд на то, почему человек отличается от других животных, при том что мы разделяем с ними большую часть генов, — говорит Мартин Тэйлор из Института генетической и молекулярной медицины Эдинбургского университета. — Сравнение атласа человека и мыши показывает, что в процессе эволюции произошел значительный перемонтаж связей между регуляторами».
— Сравнение атласа человека и мыши показывает, что в процессе эволюции произошел значительный перемонтаж связей между регуляторами».
Человек тоже различен в разных тканях. Секрет того, что клетки печени не похожи на клетки крови, опять же в том, что в них работают разные гены.
А каким генам работать, каким — нет, зависит от регуляции.
Регуляторы генов
 Сигнал к этому возникает, когда промотор узнается ферментом РНК-полимеразой. Энхансер — это умножитель работы гена, последовательность нуклеотидов, которая может располагаться вовсе не рядом с геном, а где-то довольно далеко. Но, действуя через определенные белки, энхансер может в несколько раз увеличить транскрипцию того или иного гена.
Сигнал к этому возникает, когда промотор узнается ферментом РНК-полимеразой. Энхансер — это умножитель работы гена, последовательность нуклеотидов, которая может располагаться вовсе не рядом с геном, а где-то довольно далеко. Но, действуя через определенные белки, энхансер может в несколько раз увеличить транскрипцию того или иного гена.Определить расположение в геноме этих регуляторов — промоторов и энхансеров — очень сложная задача, которая может быть решена только усилиями большого консорциума. Эта задача и решалась в программе Fantom. Например, ученые из Института Рослина Эдинбургского университета создали атлас регуляторов генной активности при развитии мышц и костей.
Исследователи использовали специальную технику под названием Cap Analysis of Gene Expression (CAGE), созданную в центре RIKEN. Она позволила проследить за активностью промоторов и энхансеров более чем в 180 типах клеток человека.
Ученым удалось идентифицировать в геноме 180 тыс. промоторов и 44 тыс.
Они нашли, что регуляция транскрипции в очень большой степени специфична для каждого типа клеток.
Российские ученые в Fantom
В консорциум проекта Fantom 5 входит команда российских биоинформатиков из Института общей генетики РАН. Об этой работе «Газете.Ru» рассказал доктор физико-математических наук Всеволод Макеев, заведующий отделом ИОГен РАН, координатор российской группы.
— Как давно вы участвуете в проекте?
— Мы присоединились к консорциуму Fantom в 2011 году, на этапе Fantom 5. Когда стало возможно получать данные для большого количества тканей, руководители проекта привлекли биоинформатиков со всего мира. Получая доступ к уникальным данным до того, как они будут опубликованы, биоинформатики, в свою очередь, предлагали методы обработки данных и идеи, которые можно проверить этими методами.
— Как организована ваша работа?
— Все данные лежат на серверах, мы их скачиваем и анализируем, и это можно делать, находясь в Москве.
Мы используем для этого мощные компьютеры на факультете биоинженерии и биоинформатики МГУ и у себя в ИОГене.
— А какие конкретные задачи перед вами стояли?
— Мы исследовали активность регуляторных участков генома и белков — транскрипционных факторов, которые с этими белками взаимодействуют. Над этим работали пять групп, причем это большая часть всех исследовательских групп в мире, которые решают эти задачи. В результате удалось составить атлас активности регуляторных белков в разных клетках — это около 1500 белков. Но районы, в которых они взаимодействуют с ДНК, известны пока менее чем для половины регуляторов. Удалось также выявить роль химического изменения ДНК, так называемого метилирования, в связывании регуляторных белков с ДНК.
Удалось также выявить роль химического изменения ДНК, так называемого метилирования, в связывании регуляторных белков с ДНК.
— Как шло развитие исследований от первого к пятому Fantom? И на каких объектах работали ученые?
— С 2000 года по крайней мере три раза менялась технология, и новые методы позволяли проводить все более широкие и точные исследования. А что касается объектов, изначально это была мышь, и буква «М» в аббревиатуре означала Mouse.
Сейчас это мышь и человек
— Чем отличается проект Fantom от проекта Encode, результаты которого не так давно были опубликованы?
— Главным образом тем, что мы анализировали не раковые линии, а нормальные клетки. С раковыми линиями проще работать, но они не всегда хорошо отражают процессы в нормальных клетках. Такой широкий анализ клеток нормальных тканей не проводился еще никогда. Благодаря этому появилась возможность понять, почему клетки тканей имеют такой клеточный тип, какой они имеют. Это важно и для регенеративной клеточной медицины — понять, как устроена дифференцировка клеток в тканях. И вообще это фундаментальный вопрос биологии.
И вообще это фундаментальный вопрос биологии.
— Есть ли уже понимание, почему клетки становятся разными и как работает геном?
— Сейчас мы более-менее понимаем, какие гены в каких клетках работают, причем это не обязательно гены, которые кодируют белки. Но более интересен вопрос, почему в какой-то ткани, допустим, включены 10 тыс. генов, а остальные не работают. Почему промоторы генов включаются и выключаются. Пока еще мы не понимаем этого настолько хорошо, чтобы мы могли этим процессом управлять, чтобы мы могли, скажем, конструировать регуляторные элементы, вставлять генно-инженерные последовательности в геном и говорить, что этот ген будет работать в таких-то тканях, а в таких-то тканях он работать не будет.
— А будет ли продолжение проекта?
— Да, сейчас опубликованы результаты первой стадии Fantom 5, а результаты второй стадии, вероятно, будут опубликованы в 2015 году. Обсуждаются и возможности дальнейшего продолжения проекта.
Осталось понять, как самолет летает
«Мы являемся многоклеточными организмами, состоящими по меньшей мере из 400 типов клеток. Это замечательное разнообразие типов клеток позволяет нам видеть, думать, слышать, двигаться и сопротивляться инфекциям, при этом вся эта информация закодирована в геноме, одном и том же у всех клеток. Различия между клетками состоят в том, какую именно часть генома они используют. Например, в клетках мозга используются гены, которые не используются в клетках печени, и поэтому мозг и печень работают по-разному.
Это замечательное разнообразие типов клеток позволяет нам видеть, думать, слышать, двигаться и сопротивляться инфекциям, при этом вся эта информация закодирована в геноме, одном и том же у всех клеток. Различия между клетками состоят в том, какую именно часть генома они используют. Например, в клетках мозга используются гены, которые не используются в клетках печени, и поэтому мозг и печень работают по-разному.
В рамках проекта Fantom 5 мы впервые выяснили, какие именно гены используются в каждой из клеток человеческого тела и какие участки генома этим управляют», — сказал доктор Алистер Форрест, сотрудник центра RIKEN, научный координатор проекта Fantom 5.
Особенность проекта в том, что он направлен на изучение нормальных, здоровых клеток, так называемых первичных, не измененных генетически, не раковых. Чтобы понять, как работает геном, нужно делать это в здоровых клетках. Но в дальнейшем эти же методы позволят изучить генную активность и в патологических клетках у пациентов с самыми разными заболеваниями, чтобы понять механизм поломки на молекулярном уровне.
Профессор Дэвид Хьюм, директор Института Рослина Эдинбургского университета, используя аналогию с самолетом, сказал: «Мы сделали скачок в понимании того, как работают отдельные детали самолета. И узнали довольно много о том, как они взаимодействуют между собой, чтобы в конечном счете понять, как самолет летает».
Загадки истории: что содержится в ДНК русского человека
https://ria.ru/20190620/1555710999.html
Загадки истории: что содержится в ДНК русского человека
Загадки истории: что содержится в ДНК русского человека — РИА Новости, 03.03.2020
Загадки истории: что содержится в ДНК русского человека
Ученые опровергли поговорку «поскреби русского — найдешь татарина». Монгольское нашествие почти не оставило следа в русских геномах, не были нашими прямыми… РИА Новости, 03.03.2020
2019-06-20T08:00
2019-06-20T08:00
2020-03-03T14:40
россия
днк
санкт-петербургский государственный университет
наука
/html/head/meta[@name=’og:title’]/@content
/html/head/meta[@name=’og:description’]/@content
https://cdn24. img.ria.ru/images/155571/14/1555711428_0:225:1250:928_1920x0_80_0_0_f9457477dc90e6dd4c904651f8ac282a.jpg
img.ria.ru/images/155571/14/1555711428_0:225:1250:928_1920x0_80_0_0_f9457477dc90e6dd4c904651f8ac282a.jpg
МОСКВА, 20 июн — РИА Новости, Татьяна Пичугина. Ученые опровергли поговорку «поскреби русского — найдешь татарина». Монгольское нашествие почти не оставило следа в русских геномах, не были нашими прямыми предками и скифы. От кого же произошли русские и что можно узнать о них по ДНК — в материале РИА Новости.Из чего состоит геном россиянина»В геноме россиянина, как в геноме любого другого организма, содержится четыре нуклеотида: аденин, гуанин, цитозин и тимин, представляющие собой моноэфиры ортофосфорной кислоты и соединенные фосфодиэфирной связью. Более 99,5 процента нуклеотидных последовательностей в геномах всех людей на Земле идентичны, а вот на эти полпроцента или даже меньше — одну десятую — приходятся все различия», — комментирует РИА Новости Владимир Брюхин, ведущий научный сотрудник Центра геномной биоинформатики имени Ф. Г. Добржанского СПбГУ. При наследовании ДНК от поколения к поколению в его структуре возникают различные изменения. Это вставки или пробелы (делеции) фрагментов, длинные или короткие повторы определенного сочетания нуклеотидов, однонуклеотидные полимормизмы, когда в каком-то участке гена заменяется всего одна буква, и другие варианты. Одни происходят случайно (генетический дрейф), другие — результат приспособления к условиям среды. Все это, как правило, находится в некодирующей части генома, той, что не несет информацию о синтезе белков.Возникший вариант генома может наследоваться и закрепиться в популяции. Тогда он служит маркером, по которому одни популяции отличают от других. При этом популяции далеко не всегда удается однозначно сопоставить с историческим народом. Ученые обнаружили большое разнообразие геномовВ России почти две сотни народностей, из которых примерно восемьдесят процентов относят себя к русской национальности. Но даже их ученые считают «полиэтносом», смесью древних балтославянских и германских племен, финно-угорских и тюркских народов, множества более мелких этносов. Геномы русских разных областей, часто соседних, заметно различаются.
Это вставки или пробелы (делеции) фрагментов, длинные или короткие повторы определенного сочетания нуклеотидов, однонуклеотидные полимормизмы, когда в каком-то участке гена заменяется всего одна буква, и другие варианты. Одни происходят случайно (генетический дрейф), другие — результат приспособления к условиям среды. Все это, как правило, находится в некодирующей части генома, той, что не несет информацию о синтезе белков.Возникший вариант генома может наследоваться и закрепиться в популяции. Тогда он служит маркером, по которому одни популяции отличают от других. При этом популяции далеко не всегда удается однозначно сопоставить с историческим народом. Ученые обнаружили большое разнообразие геномовВ России почти две сотни народностей, из которых примерно восемьдесят процентов относят себя к русской национальности. Но даже их ученые считают «полиэтносом», смесью древних балтославянских и германских племен, финно-угорских и тюркских народов, множества более мелких этносов. Геномы русских разных областей, часто соседних, заметно различаются. Словом, подвести под общий знаменатель все генетическое многообразие русских и получить некий геном «среднего россиянина» нереально.По этой причине, например, для проекта «Российские геномы», который реализуется под эгидой СПбГУ при участии ИОГен РАН, ИТМО и еще множества научных организаций страны, выбрали более пятидесяти популяций, в том числе тридцать региональных русских этнических групп. Пока секвенировали 330 геномов из 17 популяций. Этого мало для статистики, но некоторыми результатами ученые на днях поделились.»По предварительным данным, в целом у русских много общего с финно-уграми, балтийским и западноевропейскими геномами, что, впрочем, отражает историю миграции и расселения народа. Хотя общего единства пока не прослеживается: геномы псковской и новгородской популяций похожи на балтийские, архангельские почти не отличаются от западных финно-угорских, а южные русские близки западноевропейским и практически не содержат финно-угорского компонента, в отличие от русских северо-западной и центральной частей России», — продолжает ученый.
Словом, подвести под общий знаменатель все генетическое многообразие русских и получить некий геном «среднего россиянина» нереально.По этой причине, например, для проекта «Российские геномы», который реализуется под эгидой СПбГУ при участии ИОГен РАН, ИТМО и еще множества научных организаций страны, выбрали более пятидесяти популяций, в том числе тридцать региональных русских этнических групп. Пока секвенировали 330 геномов из 17 популяций. Этого мало для статистики, но некоторыми результатами ученые на днях поделились.»По предварительным данным, в целом у русских много общего с финно-уграми, балтийским и западноевропейскими геномами, что, впрочем, отражает историю миграции и расселения народа. Хотя общего единства пока не прослеживается: геномы псковской и новгородской популяций похожи на балтийские, архангельские почти не отличаются от западных финно-угорских, а южные русские близки западноевропейским и практически не содержат финно-угорского компонента, в отличие от русских северо-западной и центральной частей России», — продолжает ученый. Гены рассказывают об особенностях здоровьяИсследователей интересует как этническое происхождение, так и имеющие отношение к здоровью варианты генов: предрасположенность к заболеваниям, эффективность лекарств, возможные последствия их приема. «Как показали наши исследования, в среднем в геноме каждого человека 50-60 геномных вариантов, влияющих на вероятность развития того или иного заболевания», — отмечает Брюхин.Давно известно, что некоторые наследственные заболевания встречаются в одних популяциях чаще, чем в других. Например, фенилкетонурия, которая вызвана нарушением метаболизма и приводит при неправильном питании к задержке умственного развития, не так уж редка у европейцев и русских. А вот у марийцев, чувашей, удмуртов и адыгейцев ее почти нет. В какой степени за это отвечают генетические различия, ученым предстоит выяснить. «Распространенность генетического варианта в гене TBC1D31, ассоциированного, например, с диабетическим заболеванием почек, различается даже между псковской и новгородской популяциями почти в два раза и в семь раз по сравнению с якутской популяцией», — добавляет ученый, подчеркивая, что это предварительные данные.
Гены рассказывают об особенностях здоровьяИсследователей интересует как этническое происхождение, так и имеющие отношение к здоровью варианты генов: предрасположенность к заболеваниям, эффективность лекарств, возможные последствия их приема. «Как показали наши исследования, в среднем в геноме каждого человека 50-60 геномных вариантов, влияющих на вероятность развития того или иного заболевания», — отмечает Брюхин.Давно известно, что некоторые наследственные заболевания встречаются в одних популяциях чаще, чем в других. Например, фенилкетонурия, которая вызвана нарушением метаболизма и приводит при неправильном питании к задержке умственного развития, не так уж редка у европейцев и русских. А вот у марийцев, чувашей, удмуртов и адыгейцев ее почти нет. В какой степени за это отвечают генетические различия, ученым предстоит выяснить. «Распространенность генетического варианта в гене TBC1D31, ассоциированного, например, с диабетическим заболеванием почек, различается даже между псковской и новгородской популяциями почти в два раза и в семь раз по сравнению с якутской популяцией», — добавляет ученый, подчеркивая, что это предварительные данные. А если поскрести глубжеКак генетики увязывают ДНК и этническую принадлежность? Отправляются в экспедиции в различные регионы, берут образцы у местных и записывают, к какой национальности они себя причисляют, откуда их родители, дедушки и бабушки. Если минимум три поколения семьи проживали в одной деревне и называли себя русскими, такой геном относят к этому этносу, происходящему из определенной местности. Затем из образцов слюны или крови в лаборатории выделяют ядерные и митохондриальные ДНК и выполняют полное секвенирование. Результаты — цепочки из миллиардов букв — анализируют в программах, вычленяя известные маркеры, ища новые, и сравнивают между собой. Методы извлечения и секвенирования, как и алгоритмы анализа, постоянно совершенствуются. В 2015 году ученые Института общей генетики РАН вместе с зарубежными коллегами опубликовали результаты масштабного исследования геномов русских. По их данным, отчетливо выделяются северные, центральные и южные группы. Различие в «субстрате», то есть этносах, живших на территории европейской части России до прихода славян и балтов.
А если поскрести глубжеКак генетики увязывают ДНК и этническую принадлежность? Отправляются в экспедиции в различные регионы, берут образцы у местных и записывают, к какой национальности они себя причисляют, откуда их родители, дедушки и бабушки. Если минимум три поколения семьи проживали в одной деревне и называли себя русскими, такой геном относят к этому этносу, происходящему из определенной местности. Затем из образцов слюны или крови в лаборатории выделяют ядерные и митохондриальные ДНК и выполняют полное секвенирование. Результаты — цепочки из миллиардов букв — анализируют в программах, вычленяя известные маркеры, ища новые, и сравнивают между собой. Методы извлечения и секвенирования, как и алгоритмы анализа, постоянно совершенствуются. В 2015 году ученые Института общей генетики РАН вместе с зарубежными коллегами опубликовали результаты масштабного исследования геномов русских. По их данным, отчетливо выделяются северные, центральные и южные группы. Различие в «субстрате», то есть этносах, живших на территории европейской части России до прихода славян и балтов. Пытаться идентифицировать этот древний предковый субстрат с нынешними народами неправильно. Ученые склоняются к выводу, что он существовал еще до разделения популяций на славян, балтов, германцев, финно-угров и так далее. Нас с ним разъединяет не одно тысячелетие. Кем были эти народы, носителями каких культур, еще предстоит узнать. Распространенное мнение о том, что славяне — прямые потомки скифов и в более широком смысле — азиатов, не подтверждается по тем же причинам: скифы жили две с половиной тысячи лет назад. Их гены могут быть и у русских, но только через посредничество каких-то других, более близких к нам по времени этносов. Это как с генами неандертальцев и денисовцев, которые есть у русских, как и у большинства современных популяций людей, поскольку все мы происходим от одних предков, вышедших из Африки сотни тысяч лет назад. Ученые также отрицают большой вклад татаро-монголов в русский генофонд. Иго повлияло на историю и культуру, но в генах его след едва заметен. Азиатский компонент в небольшом количестве присутствует, но более древний, от этносов, населявших Сибирь задолго до событий XII-XIV веков.
Пытаться идентифицировать этот древний предковый субстрат с нынешними народами неправильно. Ученые склоняются к выводу, что он существовал еще до разделения популяций на славян, балтов, германцев, финно-угров и так далее. Нас с ним разъединяет не одно тысячелетие. Кем были эти народы, носителями каких культур, еще предстоит узнать. Распространенное мнение о том, что славяне — прямые потомки скифов и в более широком смысле — азиатов, не подтверждается по тем же причинам: скифы жили две с половиной тысячи лет назад. Их гены могут быть и у русских, но только через посредничество каких-то других, более близких к нам по времени этносов. Это как с генами неандертальцев и денисовцев, которые есть у русских, как и у большинства современных популяций людей, поскольку все мы происходим от одних предков, вышедших из Африки сотни тысяч лет назад. Ученые также отрицают большой вклад татаро-монголов в русский генофонд. Иго повлияло на историю и культуру, но в генах его след едва заметен. Азиатский компонент в небольшом количестве присутствует, но более древний, от этносов, населявших Сибирь задолго до событий XII-XIV веков. Один из показательных примеров — изучение геномов казачества. Некоторые историки допускают, что раз казаки жили на границе Руси, охраняя ее от набегов тюркоязычных племен, то они могли в итоге вобрать в себя степной (подразумевается монголо-татарский) компонент. Российские ученые вместе с украинскими коллегами решили это проверить и секвенировали геномы четырех казачьих групп. Выяснилось, что на девяносто процентов генофонд донских верхних и нижних, кубанских, запорожских аналогичен восточнославянскому, как у русских, украинцев, белорусов. А вот терские казаки — исключение, у них заметный вклад северокавказских генов. Исследование геномов русских и других этносов, проживающих в стране, — это мейнстрим мировой науки. Без этого невозможно установить происхождение современных популяций, древних миграций населения, уточнить и проверить исторические гипотезы. И это необходимо для того, чтобы изучить распространение наследственных заболеваний, обнаружить генетические маркеры, которые помогут сделать медицину адресной.
Один из показательных примеров — изучение геномов казачества. Некоторые историки допускают, что раз казаки жили на границе Руси, охраняя ее от набегов тюркоязычных племен, то они могли в итоге вобрать в себя степной (подразумевается монголо-татарский) компонент. Российские ученые вместе с украинскими коллегами решили это проверить и секвенировали геномы четырех казачьих групп. Выяснилось, что на девяносто процентов генофонд донских верхних и нижних, кубанских, запорожских аналогичен восточнославянскому, как у русских, украинцев, белорусов. А вот терские казаки — исключение, у них заметный вклад северокавказских генов. Исследование геномов русских и других этносов, проживающих в стране, — это мейнстрим мировой науки. Без этого невозможно установить происхождение современных популяций, древних миграций населения, уточнить и проверить исторические гипотезы. И это необходимо для того, чтобы изучить распространение наследственных заболеваний, обнаружить генетические маркеры, которые помогут сделать медицину адресной.
https://ria.ru/20180826/1527190793.html
https://ria.ru/20190417/1552774350.html
https://sn.ria.ru/20171220/1511282408.html
https://ria.ru/20190407/1552388126.html
россия
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
2019
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
Новости
ru-RU
https://ria.ru/docs/about/copyright.html
https://xn--c1acbl2abdlkab1og.xn--p1ai/
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
https://cdn25.img.ria.ru/images/155571/14/1555711428_0:108:1250:1046_1920x0_80_0_0_89468a9c2fc6211fe33267dd3482c59b.jpgРИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
РИА Новости
7 495 645-6601
ФГУП МИА «Россия сегодня»
https://xn--c1acbl2abdlkab1og.xn--p1ai/awards/
россия, днк, санкт-петербургский государственный университет
МОСКВА, 20 июн — РИА Новости, Татьяна Пичугина. Ученые опровергли поговорку «поскреби русского — найдешь татарина». Монгольское нашествие почти не оставило следа в русских геномах, не были нашими прямыми предками и скифы. От кого же произошли русские и что можно узнать о них по ДНК — в материале РИА Новости.
Из чего состоит геном россиянина
«В геноме россиянина, как в геноме любого другого организма, содержится четыре нуклеотида: аденин, гуанин, цитозин и тимин, представляющие собой моноэфиры ортофосфорной кислоты и соединенные фосфодиэфирной связью. Более 99,5 процента нуклеотидных последовательностей в геномах всех людей на Земле идентичны, а вот на эти полпроцента или даже меньше — одну десятую — приходятся все различия», — комментирует РИА Новости Владимир Брюхин, ведущий научный сотрудник Центра геномной биоинформатики имени Ф. Г. Добржанского СПбГУ.
При наследовании ДНК от поколения к поколению в его структуре возникают различные изменения. Это вставки или пробелы (делеции) фрагментов, длинные или короткие повторы определенного сочетания нуклеотидов, однонуклеотидные полимормизмы, когда в каком-то участке гена заменяется всего одна буква, и другие варианты. Одни происходят случайно (генетический дрейф), другие — результат приспособления к условиям среды. Все это, как правило, находится в некодирующей части генома, той, что не несет информацию о синтезе белков.
Возникший вариант генома может наследоваться и закрепиться в популяции. Тогда он служит маркером, по которому одни популяции отличают от других. При этом популяции далеко не всегда удается однозначно сопоставить с историческим народом.
26 августа 2018, 08:00НаукаЛихой детектив: как русские распоряжаются своим генетическим материаломУченые обнаружили большое разнообразие геномов
В России почти две сотни народностей, из которых примерно восемьдесят процентов относят себя к русской национальности. Но даже их ученые считают «полиэтносом», смесью древних балтославянских и германских племен, финно-угорских и тюркских народов, множества более мелких этносов. Геномы русских разных областей, часто соседних, заметно различаются. Словом, подвести под общий знаменатель все генетическое многообразие русских и получить некий геном «среднего россиянина» нереально.
По этой причине, например, для проекта «Российские геномы», который реализуется под эгидой СПбГУ при участии ИОГен РАН, ИТМО и еще множества научных организаций страны, выбрали более пятидесяти популяций, в том числе тридцать региональных русских этнических групп. Пока секвенировали 330 геномов из 17 популяций. Этого мало для статистики, но некоторыми результатами ученые на днях поделились.«По предварительным данным, в целом у русских много общего с финно-уграми, балтийским и западноевропейскими геномами, что, впрочем, отражает историю миграции и расселения народа. Хотя общего единства пока не прослеживается: геномы псковской и новгородской популяций похожи на балтийские, архангельские почти не отличаются от западных финно-угорских, а южные русские близки западноевропейским и практически не содержат финно-угорского компонента, в отличие от русских северо-западной и центральной частей России», — продолжает ученый.
17 апреля 2019, 14:00НаукаГенетики выяснили, что связывает жителей Пскова, Новгорода и ЯкутииГены рассказывают об особенностях здоровья
Исследователей интересует как этническое происхождение, так и имеющие отношение к здоровью варианты генов: предрасположенность к заболеваниям, эффективность лекарств, возможные последствия их приема.
«Как показали наши исследования, в среднем в геноме каждого человека 50-60 геномных вариантов, влияющих на вероятность развития того или иного заболевания», — отмечает Брюхин.
Давно известно, что некоторые наследственные заболевания встречаются в одних популяциях чаще, чем в других. Например, фенилкетонурия, которая вызвана нарушением метаболизма и приводит при неправильном питании к задержке умственного развития, не так уж редка у европейцев и русских. А вот у марийцев, чувашей, удмуртов и адыгейцев ее почти нет. В какой степени за это отвечают генетические различия, ученым предстоит выяснить.
«Распространенность генетического варианта в гене TBC1D31, ассоциированного, например, с диабетическим заболеванием почек, различается даже между псковской и новгородской популяциями почти в два раза и в семь раз по сравнению с якутской популяцией», — добавляет ученый, подчеркивая, что это предварительные данные.
20 декабря 2017, 08:00
Зов предков: откуда у новорожденных в регионах России генетические болезниА если поскрести глубже
Как генетики увязывают ДНК и этническую принадлежность? Отправляются в экспедиции в различные регионы, берут образцы у местных и записывают, к какой национальности они себя причисляют, откуда их родители, дедушки и бабушки. Если минимум три поколения семьи проживали в одной деревне и называли себя русскими, такой геном относят к этому этносу, происходящему из определенной местности.
Затем из образцов слюны или крови в лаборатории выделяют ядерные и митохондриальные ДНК и выполняют полное секвенирование. Результаты — цепочки из миллиардов букв — анализируют в программах, вычленяя известные маркеры, ища новые, и сравнивают между собой. Методы извлечения и секвенирования, как и алгоритмы анализа, постоянно совершенствуются.
В 2015 году ученые Института общей генетики РАН вместе с зарубежными коллегами опубликовали результаты масштабного исследования геномов русских. По их данным, отчетливо выделяются северные, центральные и южные группы. Различие в «субстрате», то есть этносах, живших на территории европейской части России до прихода славян и балтов.7 апреля 2019, 10:00НаукаГенописная летопись: естественнонаучная революция в истории РоссииПытаться идентифицировать этот древний предковый субстрат с нынешними народами неправильно. Ученые склоняются к выводу, что он существовал еще до разделения популяций на славян, балтов, германцев, финно-угров и так далее. Нас с ним разъединяет не одно тысячелетие. Кем были эти народы, носителями каких культур, еще предстоит узнать.
Распространенное мнение о том, что славяне — прямые потомки скифов и в более широком смысле — азиатов, не подтверждается по тем же причинам: скифы жили две с половиной тысячи лет назад. Их гены могут быть и у русских, но только через посредничество каких-то других, более близких к нам по времени этносов.
Это как с генами неандертальцев и денисовцев, которые есть у русских, как и у большинства современных популяций людей, поскольку все мы происходим от одних предков, вышедших из Африки сотни тысяч лет назад.
Ученые также отрицают большой вклад татаро-монголов в русский генофонд. Иго повлияло на историю и культуру, но в генах его след едва заметен. Азиатский компонент в небольшом количестве присутствует, но более древний, от этносов, населявших Сибирь задолго до событий XII-XIV веков.
Один из показательных примеров — изучение геномов казачества. Некоторые историки допускают, что раз казаки жили на границе Руси, охраняя ее от набегов тюркоязычных племен, то они могли в итоге вобрать в себя степной (подразумевается монголо-татарский) компонент.
Российские ученые вместе с украинскими коллегами решили это проверить и секвенировали геномы четырех казачьих групп. Выяснилось, что на девяносто процентов генофонд донских верхних и нижних, кубанских, запорожских аналогичен восточнославянскому, как у русских, украинцев, белорусов. А вот терские казаки — исключение, у них заметный вклад северокавказских генов.Исследование геномов русских и других этносов, проживающих в стране, — это мейнстрим мировой науки. Без этого невозможно установить происхождение современных популяций, древних миграций населения, уточнить и проверить исторические гипотезы. И это необходимо для того, чтобы изучить распространение наследственных заболеваний, обнаружить генетические маркеры, которые помогут сделать медицину адресной.
Гинцбург отверг возможность влияния вакцины «Спутник V» на ДНК человека :: Общество :: РБК
Вакцина «Спутник V» не влияет на наследственность, заявил глава Центра им. Гамалеи Александр Гинцбург. Она не воспроизводится в организме и никак не может встроиться в ДНК, пояснил он
Фото: Hector Vivas / Getty Images
Вакцина против коронавируса «Спутник V» никак не может влиять на геном человека или генетическую наследственность, заявил в интервью «РИА Новости» глава Центра им. Гамалеи Александр Гинцбург.
«Не может, ответ однозначный. На геном человека может влиять то, что может в геном человека интегрироваться или каким-то образом влиять на структуру ДНК. <…> Если препарат в организме человека не размножается, то он никак не может интегрироваться и взаимодействовать с нашей нуклеиновой кислотой. Поэтому «Спутник V» никак не может влиять ни на нуклеиновую кислоту, ни на наследственность», — сказал Гинцбург.
Роспотребнадзор предупредил об особенностях антител от вакцины «Вектора»«Спутник V» разрешили применять в России в августе 2020 года. Препарат зарегистрировали еще до окончания финальной фазы испытаний. Она должна завершиться к середине июня, сообщил Гинцбург.
Эта вакцина произведена на основе неопасного для человека аденовируса со встроенным участком генома коронавируса SARS-CoV-2, который и провоцирует иммунный ответ. Однако к аденовирусу у человека тоже могут выработаться антитела, и тогда повторно привиться «Спутником V» будет нельзя, поскольку эти антитела будут разрушать компоненты вакцины, допустила в марте заместитель директора по клинико-аналитической работе ЦНИИ эпидемиологии Роспотребнадзора Наталья Пшеничная.
Краткий словарь генетических терминов
Краткий словарь основных понятий и терминов, использующихся в генетике
Для понимания того, с чем работает наша компания и зачем эта работа нужна, какие результаты мы получаем и что они вам расскажут, можно прийти на консультацию к специалистам ЦГРМ «ГЕНЕТИКО». А для того, чтобы Вы не забыли, о чем был разговор, и не утонули в море новой информации, мы составили для Вас небольшой словарик основных понятий и терминов, использующихся в генетике.
Основным положением биологической науки является то, что клетка – это самое маленькое из возможных проявление жизни и что новая клетка может появиться только от уже существующей и никак не может возникнуть сама по себе. Конечно, это приводит к большому количеству вопросов о том, как зародилась жизнь и каким образом могла сформироваться самая первая клетка. Но для удобства будем считать обозначенные положения верными в современной реальности планеты Земля, где мы живем. Несмотря на невообразимо огромное разнообразие живых существ, все они состоят из клеток. И у всех клеток есть схожие черты, которые обусловлены самыми простыми жизненными необходимостями. Во-первых, клетка должна как-то отделяться от внешнего пространства – для этого есть специальная оболочка.
Во-вторых, клетка должна питаться – для этого есть разные системы, способные преобразовать энергию света или химических связей в необходимые для жизни вещества и удобную для использования энергию. И еще клетка умеет размножаться. Для выполнения всех этих функций необходимы механизмы, основу которых составляют белки и РНК. А вот инструкция, как эти молекулы должны выглядеть и работать, хранится в специальном отсеке клетки – ядре – в виде ДНК. Ошибки в этой инструкции, которая разрабатывалась миллионы лет, приводят к смерти клетки. А в многоклеточном организме, таком, как у человека, например, клетки взаимодействуют друг с другом, поэтому нарушение в работе одной или нескольких клеток может привести не к смерти всего организма, а к нарушениям его работы – заболеваниям. Также необходимо помнить, что человеческий организм огромная система, ансамбль миллионов разнообразных маленьких организмов, которые выросли из одной единственной клетки – зиготы – результата слияния яйцеклетки и сперматозоида.
ДНК – ДезоксиРибонуклиновая Кислота – полимер, то есть молекула с большим количеством последовательно повторяющихся структурных элементов, который несет всю информацию о генах и белках, необходимых для жизни всего организма. ДНК является картотекой, библиотекой и матрицей, с которой считывается информация в определенной последовательности и определенных условиях, разъяснения о которых записаны как в самой ДНК, так и с помощью различных дополнительных модификаций этой молекулы. Каждой хромосоме соответствует 1 молекула ДНК. Структурными блоками этого полимера являются дезоксирибонуклеотиды (=нуклеотиды), которые бывают 4х видов (А, Т, Г, Ц).
Последовательность ДНК – это то, в каком порядке в молекуле ДНК идут ее структурные элементы – нуклеотиды. Таким образом, генетической информацией является именно последовательность ДНК, а молекула ДНК является ее физическим носителем.
Хромосома – это молекула ДНК, специальным образом обернутая различными белками, которые помогают управляться с такой длинной молекулой, чтобы она не порвалась, не перепуталась с другими ДНК-молекулами и была физически доступна для белков, осуществляющих работу всего генетического аппарата.
РНК –РибоНуклиновая Кислота – полимер, который выполняет функциональную роль переносчика информации, то есть копии, которая делается с ДНК и используется для создания функциональных молекул: специальных РНК или белков. Специальные молекулы РНК могут не являться матрицами, на базе которых синтезируется белок, а сами выполняют структурные, ферментативные или транспортные функции. Главное, что последовательность структурных блоков в молекуле РНК всегда определена последовательностью ДНК соответствующего участка.
Белок – основная функциональная единица живой клетки с самым широчайшим спектром функций и возможностей. Как ДНК и РНК, является полимером, однако имеет химически иные структурные блоки – аминокислоты. Их последовательность, с одной стороны, напрямую зависит от соответствующей последовательности ДНК и может изменяться только в ограниченных и предусмотренных в ДНК инструкций, с другой стороны является основой структуры, в том числе пространственной, возможностей и функции белков разных типов.
Ген – определение гена включает два аспекта: теоретический и физический. Теоретически, то есть умозрительно, геном называют последовательность ДНК (слово, записанное на языке генетики), обладающее определенными свойствами. Как и слово в языке, ген является основой наследственной информации, в то время как различные другие структуры можно отнести к знакам препинания или вспомогательным элементам. Ген является подробной инструкцией для синтеза белка или специфической РНК, которую он кодирует. Причем эта инструкция описывает не только последовательность молекул, но и то в каких условиях и как они должны работать и выполнять свои функции. С физической, то есть материальной, точки зрения, ген – это часть молекулы ДНК с определенными структурными элементами. Как внутри слова есть приставка, корень, суффикс и окончание, позволяющие слову адаптироваться для каждой конкретной фразы, так и у гена есть промотор, экзоны и интроны. Первый обозначает начало гена, экзоны – это ключевая информация о последовательности РНК или белка, а интроны необходимы для регуляции и тонкой настройки работы гена в условиях разных тканей, органов и изменяющейся окружающей среды.
Экспрессия гена – это эффективность работы гена, так как для его функционирования недостаточно его наличия в геноме – с него должна считываться информация. Именно то, как часто и в каком объеме считывается информация с гена, выражают термином экспрессия.
Локус – участок молекулы ДНК, содержащий различный структурные элементы, в том числе один или несколько генов.
Геном– это последовательность всех молекул ДНК организма. Важно помнить, что в каждой клетке одного организма в норме содержатся одинаковые по количеству и последовательностям молекулы ДНК, а различается экспрессия конкретных генов.
Экзом – это последовательности ДНК экзомных участков генов, то есть так называемая основная кодирующая составляющая. Это то, с чем работает организм, в то время как остальная часть генома объясняет, как работать и в каких условиях как применять и настраивать кодирующую часть генома.
Мутация – изменение последовательности ДНК по сравнению другими клетками организма или другими представителями вида. Мутации могут возникать как из-за воздействия внешних неблагоприятных условий, так и из-за того, что наши ферменты работают пусть с редкими, но ошибками. Так как происходит физическое изменение в носителе информации – ДНК, такое изменение может передаваться из поколения в поколение.
Частота мутаций — относительное значение, показывающее у какой доли людей в геноме есть конкретная мутация. Частоту мутации можно рассчитать, как среднюю для всех людей, так и отдельно по расовым или национальным, или любы другим группам. В медицинской генетике под мутацией подразумевают изменение ДНК, которое может быть связано с каким-то заболеванием, и противопоставляют ее полиморфизму. Хотя по общей логике полиморфизм – это частный случай мутации.
Полиморфизм – нейтральная, а точнее безвредная, мутация, которая сравнительно часто встречается у какой-то группы организмов одного вида. Некоторые мутации встречаются часто у всех людей, некоторые – только среди представителей определенных рас или народностей.
Аллель – вариант последовательности гена в разном виде: от различия в одной букве последовательности до отсутствия целого куска последовательности или вставке лишнего. Эти различия возникают из-за мутации, которая могла произойти у далекого предка и передаться потомству через поколения. Таким образом, каждый ген у отдельного человека может быть представлен конкретным вариантом – аллелем. Для понимания аллелизма необходимо объяснить, что, например, различия в цвете глаз, волос, росте, чувствительности к алкоголю объясняются именно разными аллельными состояниями соответствующих генов.
Генотип – это все гены конкретной особи с указанием аллельного состояния каждого гена и наличия/отсутствия мутаций в межгенных участках ДНК.
Доминантный аллель. В геноме человека содержится по 2 копии каждой хромосомы. Это означает, что в каждом геноме есть две очень похожие по длине и последовательности генов молекулы ДНК, которые отличаются аллельными состояниями генов и мутациями/полиморфизмами в межгенных участках этих молекул ДНК. Из этого следует, что и каждый ген представлен в геноме 2 копиями, каждая из которых может быть определенным вариантом (аллелем) этого гена. Доминантным аллелем называется тот, одной копии которого достаточно для проявления его особенностей. То есть если хотя бы на одной из хромосом ген находится в состоянии доминантного аллеля, то ген будет работать по тому варианту, который описывается именно этим аллелем. Важно, что так как у одного гена может быть более двух вариантов (аллелей), то и доминантность аллеля определяется по отношению к каждому из вариантов, хотя есть и те, которые доминантны по сравнению со всеми другими. Встречаются варианты с одинаковой предпочтительностью для работы, тогда проявляется совместное влияние этих вариантов.
Рецессивный аллель – по аналогии с доминантным аллелем, это такое состояние гена, которое наименее предпочтительно для проявления. Поэтому если в геноме есть другая копия гена, доминантная, то задавать темп работы гена будет именно она, но если и вторая копия гена представлена рецессивным аллелем, то будет работать этот, хотя менее предпочтительный, но в такой ситуации единственно имеющийся вариант. Хотя в большинстве случаев связанные с возникновением заболевания аллели рецессивны, это вредность/полезность не является единственным определяющим фактором рецессивности/доминантности аллеля.
Гомозигота. Гомозиготой по определенной мутации/полиморфизму/аллелю называют такую клетку или организм, в генотипе которой/которого обе копии гена на двух хромосомах представлены одним вариантом, то есть не отличаются по этой мутации/полиморфизму/аллелю.
Гетерозигота. Гетерозиготой по определенной мутации/полиморфизму/аллелю называют такую клетку или организм, в генотипе которой/которого две копии гена на двух хромосомах представлены разными вариантами, то есть отличаются по этой мутации/полиморфизму/аллелю.
Секвенирование – это группа методов, позволяющая узнать последовательность нуклеотидов в молекуле ДНК. Этот метод обладает некоторыми особенностями. Во-первых, пока что ни один способ секвенирования не позволяет прочитать всю последовательность одной хромосомы, чтение идет сравнительно небольшими отрезка от 50 до несколько тысяч нуклеотидов. Во-вторых, почти все методы устроены так, что из кусочка ДНК делается много одинаковых и читаются они все. Эта особенность проявляется в таком параметре секвенирования, как глубина секвенирования, обозначаемая 10Х, 20Х, 50Х. Чем больше это значение, тем больше раз прочитан один и тот же кусок молекулы, тем точнее можно выявить ошибки секвенирования и особенности участка, например, его гетерозиготность по какой-либо мутации/полиморфизму.
Гаплотип — совокупность состояний/вариантов определенных локусов, которые расположены на одной хромосоме, и вследствие структурных особенностей эти состояния всегда наследуются вместе. То есть, например, если в одном локусе (1) гаплотипа имеется мутация (1А), а в другом (2) имеется уже другая мутация (2M), то именно в таком составе они будут наследоваться (1А2М), а смешанных вариантов (1B2M или 1A2N) не бывает или они относятся к другому гаплотипу.
Гаплогруппа — совокупность особей, имеющих сходный гаплотип по определенным локусам, которые задаются в соответствии с тем, какую задачу нужно решить, определяя гаплогруппу
Митохондриальная ДНК. Если разбираться подробнее и глубже, то генетическая информация одного человека находится не только в 46 хромосомах, располагающихся в специальном отсеке клетки – ядре, но и в клеточных органах митохондриях. У митохондрий в клетке своя задача – преобразовывать энергию, заключенную в химической связи определенных атомов, в более удобную для клетки, то есть они готовят эффективные питательные запасы из разного сырья. Митохондрии довольно сложны, их оболочка хитро устроена, чтобы опасные побочные продукты готовки не могли попасть в остальную часть клетки, поэтому все время таскать туда нужные для их работы белки не слишком продуктивно. Таким образом, у них есть своя ДНК, которая несет информацию о разных особенных белках и РНК, которые нужны именно для работы митохондрии. Такую ДНК называют митохондриальной и она является неотъемлемой и обязательной частью нашего генотипа. Передается она только от мамы, так как сперматозоид для возможности быстро перемещаться и долго оставаться живым несет самый минимум необходимой генетической информации – 23 хромосомы. А вот яйцеклетка, которой для выполнения основной функции не нужно находится в агрессивной окружающей среде, может позволить себе бОльшую массу и дополнительные запасы в виде готовых к работе станций приготовления питания – митохондрий и заранее синтезированных белков и РНК.
Гены половой дифференцировки – группа генов, играющая ведущую роль в определении будет эмбрион развиваться как девочка или как мальчик. В геноме человека основой проявления мужских или женских половых признаков является наличие/отсутствие половой хромосомы Y, а именно особо локуса этой хромосомы – SRY (Sex-determining Region on the Y chromosome). Важно отметить, что нарушения в этом локусе могут приводить не к внешним проявлениям, а к сниженной репродуктивной способности мужчины или ее полному отсутствию. Процесс дифференцировки пола у человека можно представить тремя стадиями: 1) какой набор хромосом получается при слиянии яйцеклетки (всегда несет хромосому X) и сперматозоида (с хромосомой X или Y), 2) формирование женских или мужских половых органов в зависимости от работы генов локуса SRY, 3) развитие вторичных половых органов в соответствии с типом половых органов. Нарушения на разных этапах приводят к разным проявлениям и разным заболеваниям.
Локус AZF – это участок Y-хромосомы, на котором располагаются так называемые факторы азооспермии (AZF — AZoospermia Factors). Это особые участки, которые названы так, потому что если какой-то из них отсутствует из-за мутации, то развивается азооспермия (отсутствие сперматозоидов) или олигозооспермия (малое количество сперматозоидов). Всего обнаружено три таких фактора AZFa, AZFb и AZFc. В норме наличие всех трех является минимальным необходимым условием нормального формирования сперматозоидов. Если в геноме отсутствует один из AZFa и AZFb или оба, то нарушается созревание сперматозоидов и, как следствие, полностью отсутствует репродуктивная функция. При отсутствии локуса AZFc нарушения могут быть не столь сильными, поэтому деторождение остается возможным в некоторых случаях.
Хромосомные аномалии – это крупные мутации, которые связаны с изменением последовательности ДНК не в рамках отдельного гена или нескольких, а в масштабе хромосомы или генома. Например, отсутствие (делеция) большой части или всей хромосомы, лишняя хромосома, или часть одной хромосомы соединена с частью другой хромосомы и т.д.
Наследственное заболевание – это заболевание, вызванное нарушениями в геноме, то есть мутациями, которые либо мешают формированию нормального белка (так как ген – инструкция по его построению – поврежден), либо изменяют регуляцию, то есть условия, когда, в каком месте или с кем такой белок или ген должен работать.
Моногенное заболевание – это наследственное заболевание, вызванное мутацией в одном только в одном гене. Несмотря на то, что все остальные почти 30000 генов могут быть в порядке, изменение последовательности ДНК в этом гене вызывает нарушения функционирования всего организма.
Хромосомное заболевание – наследственное заболевание, вызванное хромосомными аномалиями.
Носительство мутации – это состояние гетерозиготы по аллелю, обладающему какими-то негативными клиническими проявлениями, если он находится в геноме в виде гомозиготы.
Пробанд – человек, с которого начинается составление генеалогического дерева (родословной). Обычно пробанд – это носитель или пациент с наследственным заболеванием, проявление которого и вызвало необходимость генеалогического анализа.
Сиблинг – в генетике таким термином обозначают потомков одних родителей, то есть братьев и сестер, но не близнецов.
Автор: Жикривецкая Светлана
Биолог-исследователь
46 — норма?. Считаем хромосомы: сколько человеку для счастья нужно
Прожиточный оптимум
Сначала договоримся о терминологии. Окончательно человеческие хромосомы посчитали чуть больше полувека назад — в 1956 году. С тех пор мы знаем, что в соматических, то есть не половых клетках, их обычно 46 штук — 23 пары.
Хромосомы в паре (одна получена от отца, другая — от матери) называют гомологичными. На них расположены гены, выполняющие одинаковые функции, однако нередко различающиеся по строению. Исключение составляют половые хромосомы — Х и Y, генный состав которых совпадает не полностью. Все остальные хромосомы, кроме половых, называют аутосомами.
Количество наборов гомологичных хромосом — плоидность — в половых клетках равно одному, а в соматических, как правило, двум.
Интересно, что не у всех видов млекопитающих число хромосом постоянно. Например, у некоторых представителей грызунов, собак и оленей обнаружили так называемые В-хромосомы. Это небольшие дополнительные хромосомы, в которых практически нет участков, кодирующих белки, а делятся и наследуются они вместе с основным набором и, как правило, не влияют на работу организма. Полагают, что В-хромосомы — это просто удвоенные фрагменты ДНК, «паразитирующие» на основном геноме.
У человека до сих пор В-хромосомы обнаружены не были. Зато иногда в клетках возникает дополнительный набор хромосом — тогда говорят о полиплоидии, а если их число не кратно 23 — об анеуплоидии. Полиплоидия встречается у отдельных типов клеток и способствует их усиленной работе, в то время как анеуплоидия обычно свидетельствует о нарушениях в работе клетки и нередко приводит к ее гибели.
Делиться надо честно
Чаще всего неправильное количество хромосом является следствием неудачного деления клеток. В соматических клетках после удвоения ДНК материнская хромосома и ее копия оказываются сцеплены вместе белками когезинами. Потом на их центральные части садятся белковые комплексы кинетохоры, к которым позже прикрепляются микротрубочки. При делении по микротрубочкам кинетохоры разъезжаются к разным полюсам клетки и тянут за собой хромосомы. Если сшивки между копиями хромосомы разрушатся раньше времени, то к ним могут прикрепиться микротрубочки от одного и того же полюса, и тогда одна из дочерних клеток получит лишнюю хромосому, а вторая останется обделенной.
Деление при образовании половых клеток (мейоз) устроено более сложно. После удвоения ДНК каждая хромосома и ее копия, как обычно, сшиты когезинами. Затем гомологичные хромосомы (полученные от отца и матери), а точнее их пары, тоже сцепляются друг с другом, и получается так называемая тетрада, или четверка. А дальше клетке предстоит поделиться два раза. В ходе первого деления расходятся гомологичные хромосомы, то есть дочерние клетки содержат пары одинаковых хромосом. А во втором делении эти пары расходятся, и в результате половые клетки несут одинарный набор хромосом.
Мейоз тоже нередко проходит с ошибками. Проблема в том, что конструкция из сцепленных двух пар гомологичных хромосом может перекручиваться в пространстве или разделяться в неположенных местах. Результатом снова будет неравномерное распределение хромосом. Иногда половой клетке удается это отследить, чтобы не передавать дефект по наследству. Лишние хромосомы часто неправильно уложены или разорваны, что запускает программу гибели. Например, среди сперматозоидов действует такой отбор по качеству. А вот яйцеклеткам повезло меньше. Все они у человека образуются еще до рождения, готовятся к делению, а потом замирают. Хромосомы уже удвоены, тетрады образованы, а деление отложено. В таком виде они живут до репродуктивного периода. Дальше яйцеклетки по очереди созревают, делятся первый раз и снова замирают. Второе деление происходит уже сразу после оплодотворения. И на этом этапе проконтролировать качество деления уже сложно. А риски больше, ведь четыре хромосомы в яйцеклетке остаются сшитыми в течение десятков лет. За это время в когезинах накапливаются поломки, и хромосомы могут спонтанно разделяться. Поэтому чем старше женщина, тем больше вероятность неправильного расхождения хромосом в яйцеклетке.
Анеуплоидия в половых клетках неизбежно ведет к анеуплоидии зародыша. При оплодотворении здоровой яйцеклетки с 23 хромосомами сперматозоидом с лишней или недостающей хромосомами (или наоборот) число хромосом у зиготы, очевидно, будет отлично от 46. Но даже если половые клетки здоровы, это не дает гарантий здорового развития. В первые дни после оплодотворения клетки зародыша активно делятся, чтобы быстро набрать клеточную массу. Судя по всему, в ходе быстрых делений нет времени проверять корректность расхождения хромосом, поэтому могут возникнуть анеуплоидные клетки. И если произойдет ошибка, то дальнейшая судьба зародыша зависит от того, в каком делении это случилось. Если равновесие нарушено уже в первом делении зиготы, то весь организм вырастет анеуплоидным. Если же проблема возникла позже, то исход определяется соотношением здоровых и аномальных клеток.
Часть последних может дальше погибнуть, и мы никогда не узнаем об их существовании. А может принять участие в развитии организма, и тогда он получится мозаичным — разные клетки будут нести разный генетический материал. Мозаицизм доставляет немало хлопот пренатальным диагностам. Например, при риске рождения ребенка с синдромом Дауна иногда извлекают одну или несколько клеток зародыша (на той стадии, когда это не должно представлять опасности) и считают в них хромосомы. Но если зародыш мозаичен, то такой метод становится не особенно эффективным.
Третий лишний
Все случаи анеуплоидии логично делятся на две группы: недостаток и избыток хромосом. Проблемы, возникающие при недостатке, вполне ожидаемы: минус одна хромосома означает минус сотни генов.
Расположение хромосом в ядре клетки человека (хромосомные территории). Изображение: Bolzer et al., 2005 / Wikimedia Commons / CC BY 2.5Если гомологичная хромосома работает нормально, то клетка может отделаться только недостаточным количеством закодированных там белков. Но если среди оставшихся на гомологичной хромосоме генов какие-то не работают, то соответствующих белков в клетке не появится совсем.
В случае избытка хромосом все не так очевидно. Генов становится больше, но здесь — увы — больше не значит лучше.
Во-первых, лишний генетический материал увеличивает нагрузку на ядро: дополнительную нить ДНК нужно разместить в ядре и обслужить системами считывания информации.
Ученые обнаружили, что у людей с синдромом Дауна, чьи клетки несут дополнительную 21-ю хромосому, в основном нарушается работа генов, находящихся на других хромосомах. Видимо, избыток ДНК в ядре приводит к тому, что белков, поддерживающих работу хромосом, не хватает на всех.
Во-вторых, нарушается баланс в количестве клеточных белков. Например, если за какой-то процесс в клетке отвечают белки-активаторы и белки-ингибиторы и их соотношение обычно зависит от внешних сигналов, то дополнительная доза одних или других приведет к тому, что клетка перестанет адекватно реагировать на внешний сигнал. И наконец, у анеуплоидной клетки растут шансы погибнуть. При удвоении ДНК перед делением неизбежно возникают ошибки, и клеточные белки системы репарации их распознают, чинят и запускают удвоение снова. Если хромосом слишком много, то белков не хватает, ошибки накапливаются и запускается апоптоз — программируемая гибель клетки. Но даже если клетка не погибает и делится, то результатом такого деления тоже, скорее всего, станут анеуплоиды.
Жить будете
Если даже в пределах одной клетки анеуплоидия чревата нарушениями работы и гибелью, то неудивительно, что целому анеуплоидному организму выжить непросто. На данный момент известно только три аутосомы — 13, 18 и 21-я, трисомия по которым (то есть лишняя, третья хромосома в клетках) как-то совместима с жизнью. Вероятно, это связано с тем, что они самые маленькие и несут меньше всего генов. При этом дети с трисомией по 13-й (синдром Патау) и 18-й (синдром Эдвардса) хромосомам доживают в лучшем случае до 10 лет, а чаще живут меньше года. И только трисомия по самой маленькой в геноме, 21-й хромосоме, известная как синдром Дауна, позволяет жить до 60 лет.
Совсем редко встречаются люди с общей полиплоидией. В норме полиплоидные клетки (несущие не две, а от четырех до 128 наборов хромосом) можно обнаружить в организме человека, например в печени или красном костном мозге. Это, как правило, большие клетки с усиленным синтезом белка, которым не требуется активное деление.
Дополнительный набор хромосом усложняет задачу их распределения по дочерним клеткам, поэтому полиплоидные зародыши, как правило, не выживают. Тем не менее описано около 10 случаев, когда дети с 92 хромосомами (тетраплоиды) появлялись на свет и жили от нескольких часов до нескольких лет. Впрочем, как и в случае других хромосомных аномалий, они отставали в развитии, в том числе и умственном. Однако многим людям с генетическими аномалиями приходит на помощь мозаицизм. Если аномалия развилась уже в ходе дробления зародыша, то некоторое количество клеток могут остаться здоровыми. В таких случаях тяжесть симптомов снижается, а продолжительность жизни растет.
Гендерные несправедливости
Однако есть и такие хромосомы, увеличение числа которых совместимо с жизнью человека или даже проходит незаметно. И это, как ни удивительно, половые хромосомы. Причиной тому — гендерная несправедливость: примерно у половины людей в нашей популяции (девочек) Х-хромосом в два раза больше, чем у других (мальчиков). При этом Х-хромосомы служат не только для определения пола, но и несут более 800 генов (то есть в два раза больше, чем лишняя 21-я хромосома, доставляющая немало хлопот организму). Но девочкам приходит на помощь естественный механизм устранения неравенства: одна из Х-хромосом инактивируется, скручивается и превращается в тельце Барра. В большинстве случаев выбор происходит случайно, и в ряде клеток в результате активна материнская Х-хромосома, а в других — отцовская. Таким образом, все девочки оказываются мозаичными, потому что в разных клетках работают разные копии генов. Классическим примером такой мозаичности являются черепаховые кошки: на их Х-хромосоме находится ген, отвечающий за меланин (пигмент, определяющий, среди прочего, цвет шерсти). В разных клетках работают разные копии, поэтому окраска получается пятнистой и не передается по наследству, так как инактивация происходит случайным образом.
Кошка черепахового окраса. Фото: Lisa Ann Yount / Flickr / Public domainВ результате инактивации в клетках человека всегда работает только одна Х-хромосома. Этот механизм позволяет избежать серьезных неприятностей при Х-трисомии (девочки ХХХ) и синдромах Шерешевского — Тернера (девочки ХО) или Клайнфельтера (мальчики ХХY). Таким рождается примерно один из 400 детей, но жизненные функции в этих случаях обычно не нарушены существенно, и даже бесплодие возникает не всегда. Сложнее бывает тем, у кого хромосом больше трех. Обычно это значит, что хромосомы не разошлись дважды при образовании половых клеток. Случаи тетрасомии (ХХХХ, ХХYY, ХХХY, XYYY) и пентасомии (XXXXX, XXXXY, XXXYY, XXYYY, XYYYY) встречаются редко, некоторые из них описаны всего несколько раз за всю историю медицины. Все эти варианты совместимы с жизнью, и люди часто доживают до преклонных лет, при этом отклонения проявляются в аномальном развитии скелета, дефектах половых органов и снижении умственных способностей. Что характерно, дополнительная Y-хромосома сама по себе влияет на работу организма несильно. Многие мужчины c генотипом XYY даже не узнают о своей особенности. Это связано с тем, что Y-хромосома сильно меньше Х и почти не несет генов, влияющих на жизнеспособность.
У половых хромосом есть и еще одна интересная особенность. Многие мутации генов, расположенных на аутосомах, приводят к отклонениям в работе многих тканей и органов. В то же время большинство мутаций генов на половых хромосомах проявляется только в нарушении умственной деятельности. Получается, что в существенной степени половые хромосомы контролируют развитие мозга. На основании этого некоторые ученые высказывают гипотезу, что именно на них лежит ответственность за различия (впрочем, не до конца подтвержденные) между умственными способностями мужчин и женщин.
Кому выгодно быть неправильным
Несмотря на то что медицина знакома с хромосомными аномалиями давно, в последнее время анеуплоидия продолжает привлекать внимание ученых. Оказалось, что более 80% клеток опухолей содержат необычное количество хромосом. С одной стороны, причиной этому может служить тот факт, что белки, контролирующие качество деления, способны его затормозить. В опухолевых клетках часто мутируют эти самые белки-контролеры, поэтому снимаются ограничения на деление и не работает проверка хромосом. С другой стороны, ученые полагают, что это может служить фактором отбора опухолей на выживаемость. Согласно такой модели, клетки опухоли сначала становятся полиплоидными, а дальше в результате ошибок деления теряют разные хромосомы или их части. Получается целая популяция клеток с большим разнообразием хромосомных аномалий. Большинство из них нежизнеспособны, но некоторые могут случайно оказаться успешными, например если случайно получат дополнительные копии генов, запускающих деление, или потеряют гены, его подавляющие. Однако если дополнительно стимулировать накопление ошибок при делении, то клетки выживать не будут. На этом принципе основано действие таксола — распространенного лекарства от рака: он вызывает системное нерасхождение хромосом в клетках опухоли, которое должно запускать их программируемую гибель.
Получается, что каждый из нас может оказаться носителем лишних хромосом, по крайней мере в отдельных клетках. Однако современная наука продолжает разрабатывать стратегии борьбы с этими нежеланными пассажирами. Одна из них предлагает использовать белки, отвечающие за Х-хромосому, и натравить, например, на лишнюю 21-ю хромосому людей с синдромом Дауна. Сообщается, что на клеточных культурах этот механизм удалось привести в действие. Так что, возможно, в обозримом будущем опасные лишние хромосомы окажутся укрощены и обезврежены.
Полина Лосева
Обнаружение генотипов, лежащих в основе фенотипов человека: прошлые успехи при менделевской болезни, будущие подходы к комплексной болезни
Ботштейн, Д., Уайт, Р.Л., Сколник, М. и Дэвис, Р.В. Построение карты генетических связей у человека с использованием рестрикционного фрагмента полиморфизмы длины. г. J. Hum. Genet. 32 , 314–331 (1980).
CAS PubMed PubMed Central Google Scholar
Федер, Дж.N. et al. Новый ген, подобный MHC класса I, мутирован у пациентов с наследственным гемохроматозом. Nat. Genet. 13 , 399–408 (1996).
CAS PubMed Google Scholar
Dreyer, S.D. и другие. Мутации в LMX1B вызывают аномальный скелетный паттерн и почечную дисплазию при синдроме ногтевой надколенника. Nat. Genet. 19 , 47–50 (1998).
CAS PubMed Google Scholar
Энаттах, Н.S. et al. Выявление варианта, связанного с гиполактазией взрослого типа. Nat. Genet. 30 , 233–237 (2002).
CAS PubMed Google Scholar
Royer-Pokora, B. et al. Клонирование гена наследственного заболевания человека — хронической гранулематозной болезни — на основе его хромосомного расположения. Nature 322 , 32–38 (1986).
CAS PubMed Google Scholar
Кениг, М.и другие. Полное клонирование мышечной дистрофии Дюшенна (МДД). кДНК и предварительная геномная организация гена DMD у нормальных и больных людей. Cell 50 , 509–517 (1987).
CAS Google Scholar
Kerem, B. et al. Идентификация гена муковисцидоза: генетический анализ. Science 245 , 1073–1080 (1989).
CAS PubMed PubMed Central Google Scholar
Риордан, Дж.R. et al. Идентификация гена муковисцидоза: клонирование и характеристика комплементарной ДНК. Science 245 , 1066–1073 (1989).
CAS Google Scholar
Strathdee, C.A., Gavish, H., Shannon, W.R. & Buchwald, M. Клонирование кДНК для анемии Фанкони путем функциональной комплементации. Nature 356 , 763–767 (1992).
CAS PubMed Google Scholar
Савицкий, К.и другие. Один ген атаксии телеангиэктазии с продуктом, подобным киназе PI-3. Наука 268 , 1749–1753 (1995).
CAS PubMed Google Scholar
Wallace, M.R. et al. Ген нейрофиброматоза 1 типа: идентификация большого транскрипта, нарушенного у трех пациентов с NF1. Наука 249 , 181–186 (1990).
CAS PubMed Google Scholar
Фунг, Ю.-K.T. и другие. Структурные доказательства подлинности гена ретинобластомы человека. Наука 236 , 1657–1661 (1987).
CAS PubMed Google Scholar
Miki, Y. et al. Сильный кандидат на ген предрасположенности к раку груди и яичников BRCA1 . Science 266 , 66–71 (1994).
CAS PubMed Google Scholar
Вустер, Р.и другие. Идентификация гена предрасположенности к раку груди BRCA2 . Nature 378 , 789–792 (1995).
CAS PubMed Google Scholar
Nishisho, I. et al. Мутации генов хромосомы 5q21 у пациентов с FAP и колоректальным раком. Science 253 , 665–669 (1991).
CAS PubMed Google Scholar
Гуселла, Дж.F. et al. Полиморфный ДНК-маркер, генетически связанный с болезнью Хантингтона. Nature 306 , 234–238 (1983).
CAS PubMed Google Scholar
Группа совместных исследований болезни Хантингтона. Новый ген, содержащий тринуклеотидный повтор, который разрастается и нестабилен на хромосомах болезни Хантингтона. Cell 72 , 971–983 (1993).
Вебер, Дж.Л. и Мэй, П. Обильный класс полиморфизмов ДНК человека, которые можно типировать с помощью полимеразной цепной реакции. г. J. Hum. Genet. 44 , 388–396 (1989).
CAS PubMed PubMed Central Google Scholar
Litt, M. & Luty, J.A. Гипервариабельный микросателлит, обнаруженный с помощью in vitro амплификации динуклеотидного повтора в гене актина сердечной мышцы. г. Дж.Гм. Genet. 44 , 397–401 (1989).
CAS PubMed PubMed Central Google Scholar
Venter, J.C. et al. Последовательность генома человека. Наука 291 , 1304–1351 (2001).
CAS Google Scholar
Sachidanandam, R. et al. Карта вариаций последовательности генома человека, содержащая 1,42 миллиона однонуклеотидных полиморфизмов. Nature 409 , 928–933 (2001).
CAS PubMed PubMed Central Google Scholar
Lander, E.S. & Ботштейн, Д. Стратегии изучения гетерогенных генетических признаков у людей с использованием карты сцепления полиморфизмов длины рестрикционных фрагментов. Proc. Natl. Акад. Sci. США 83 , 7353–7357 (1986).
CAS PubMed Google Scholar
Холл, Дж.M. et al. Связь семейного рака груди с ранним началом с хромосомой 17q21. Science 250 , 1684–1689 (1990).
CAS PubMed PubMed Central Google Scholar
Lander, E.S. & Ботштейн, Д. Картирование гомозиготности — способ сопоставить рецессивные черты человека с ДНК инбредных детей. Наука 236 , 1567–1570 (1987).
CAS PubMed PubMed Central Google Scholar
Гшвенд, М.и другие. Локус анемии Фанкони на 16q определяется картированием гомозиготности. г. J. Hum. Genet. 59 , 377–384 (1996).
CAS PubMed PubMed Central Google Scholar
Саар К. и др. Локализация гена анемии Фанкони на хромосоме 9р. евро. J. Hum. Genet. 6 , 501–508 (1998).
CAS PubMed Google Scholar
Waisfisz, Q.и другие. Ген анемии Фанкони группы E, FANCE , отображается на хромосоме 6p. г. J. Hum. Genet. 64 , 1400–1405 (1999).
CAS PubMed PubMed Central Google Scholar
Bolino, A. et al. Локализация гена, ответственного за аутосомно-рецессивную демиелинизирующую невропатию с фокально свернутыми миелиновыми оболочками на хромосоме 11q23, путем картирования гомозиготности и общего гаплотипа. Hum.Мол. Genet. 5 , 1051–1054 (1996).
CAS PubMed Google Scholar
LeGuern, E. et al. Картирование гомозиготности аутосомно-рецессивной формы демиелинизирующей болезни Шарко-Мари-Тута по хромосоме 5q23-q33. Hum. Мол. Genet. 5 , 1685–1688 (1996).
CAS PubMed Google Scholar
Бухуш, А.и другие. Локус аксональной формы аутосомно-рецессивной болезни Шарко-Мари-Тута отображается на хромосоме 1q21.2q21.3 Am. J. Hum. Genet. 65 , 722–727 (1999).
CAS PubMed PubMed Central Google Scholar
Rogers, T. et al. Новый локус аутосомно-рецессивной периферической невропатии в области EGR2 на 10q23. г. J. Hum. Genet. 67 , 664–671 (2000).
CAS PubMed PubMed Central Google Scholar
Леал, А.и другие. Второй локус аксональной формы аутосомно-рецессивной болезни Шарко-Мари-Тута отображается на хромосоме 19q13.3. г. J. Hum. Genet. 68 , 269–274 (2001).
CAS PubMed PubMed Central Google Scholar
Лифтон Р.П., Гарави А.Г. и Геллер Д.С. Молекулярные механизмы гипертонии человека. Cell 104 , 545–556 (2001).
CAS PubMed Google Scholar
Hastbacka, J.и другие. Картирование неравновесия сцепления в изолированных популяциях основателей — диастрофическая дисплазия в Финляндии. Nat. Genet. 2 , 204–211 (1992).
CAS PubMed Google Scholar
Озелиус, Л.Дж. и др. Сильная аллельная ассоциация между геном торсионной дистонии (DYT1) и локусами хромосомы 9q34 у евреев ашкенази. г. J. Hum. Genet. 50 , 619–628 (1992).
CAS PubMed PubMed Central Google Scholar
Макдональд, М.E. et al. Кандидатская область болезни Хантингтона демонстрирует множество различных гаплотипов. Nat. Genet. 1 , 99–103 (1992).
CAS PubMed Google Scholar
Klein, C. et al. Поиск гаплотипа основателя PARK3 в большой когорте пациентов с болезнью Паркинсона из северной Германии. Ann. Гм. Genet. 63 , 285–291 (1999).
CAS PubMed Google Scholar
Сервис, С.К., Ланг, Д.В., Фреймер, Н.Б. & Sandkuijl, L.A. Картирование генов болезней с неравновесным сцеплением путем реконструкции предковых гаплотипов в популяциях основателей. г. J. Hum. Genet. 64 , 1728–1738 (1999).
CAS PubMed PubMed Central Google Scholar
МакПик, М.С. И Strahs, A. Оценка неравновесия сцепления по распаду общих гаплотипов, с применением к мелкомасштабному картированию. г. J. Hum. Genet. 65 , 858–875 (1999).
CAS PubMed PubMed Central Google Scholar
Morris, A.P. & Whittaker, J.C. Тонкомасштабное ассоциативное картирование локусов болезней с использованием симплексных семейств. Ann. Гм. Genet. 64 , 223–237 (2000).
CAS PubMed Google Scholar
Lam, J.C., Родер, К. и Девлин, Б. Тонкое отображение гаплотипов с помощью эволюционных деревьев. г. J. Hum. Genet. 66 , 659–673 (2000).
CAS PubMed PubMed Central Google Scholar
Liu, J.S. и другие. Байесовский анализ гаплотипов для картирования неравновесия по сцеплению. Genome Res. 11 , 1716–24 (2001).
CAS PubMed PubMed Central Google Scholar
Браунштейн, Б.H. et al. Выделение однокопийных генов человека из библиотеки клонов искусственных хромосом дрожжей. Science 244 , 1348–1351 (1989).
CAS PubMed Google Scholar
Cox, D.R. и другие. Радиационное гибридное картирование: генетический метод соматических клеток для построения карт хромосом млекопитающих с высоким разрешением. Science 250 , 245–250 (1990).
CAS PubMed Google Scholar
Кравчак, М.и другие. База данных мутаций генов человека — биомедицинский информационный и исследовательский ресурс. Hum. Мутат. 15 , 45–51 (2000).
CAS PubMed Google Scholar
Grantham, R. Формула разности аминокислот для объяснения эволюции белков. Наука 185 , 862–864 (1974).
CAS PubMed Google Scholar
Кравчак, М., Болл, Э. И Купер, Д.Н.Влияние соседних нуклеотидов на скорость замен одной пары оснований зародышевой линии в генах человека. г. J. Hum. Genet. 63 , 474–488 (1998).
CAS PubMed PubMed Central Google Scholar
Stephens, J.C. et al. Вариации гаплотипов и неравновесие по сцеплению в 313 генах человека. Science 293 , 489–493 (2001).
CAS Google Scholar
Миллер, М.П. и Кумар С. Понимание мутаций болезней человека с помощью межвидовой генетической изменчивости. Hum. Мол. Genet. 10 , 2319–2328 (2001).
CAS PubMed Google Scholar
Gillard, E.F. et al. Молекулярный и фенотипический анализ пациентов с делециями в богатой делециями области гена мышечной дистрофии Дюшенна (МДД). г. J. Hum. Genet. 45 , 507–520 (1989).
CAS PubMed PubMed Central Google Scholar
Мията Т., Миядзава С. и Ясунага Т. Два типа аминокислотных замен в эволюции белков. J. Mol. Evol. 12 , 219–236 (1979).
CAS PubMed Google Scholar
Риш Н. Гемохроматоз, HFE и генетическая сложность. Nat. Genet. 17 , 375–376 (1997).
CAS PubMed Google Scholar
Грабовски Г.А. Болезнь Гоше: частота генов и корреляции генотип / фенотип. Genet. Контрольная работа. 1 , 5–12 (1997).
CAS PubMed Google Scholar
Palzkill, T. & Botstein, D. Исследование структуры и функции β-лактамаз с использованием мутагенеза случайной замены. Proteins Struct.Funct. Genet. 14 , 29–44 (1992).
CAS PubMed Google Scholar
Риш Н. и Мерикангас К. Будущее генетических исследований сложных заболеваний человека. Наука 273 , 1516–1517 (1996).
CAS PubMed PubMed Central Google Scholar
Lander, E.S. Новая геномика: глобальные взгляды на биологию. Наука 274 , 536–539 (1996).
CAS Google Scholar
Риш Н. Поиск генетических детерминант в новом тысячелетии. Nature 405 , 847–856 (2000).
CAS PubMed PubMed Central Google Scholar
Peltonen, L. & McKusick, V.A. Рассмотрение болезней человека в постгеномную эпоху. Наука 291 , 1224–1228 (2001).
CAS PubMed PubMed Central Google Scholar
Marth, G. et al. Однонуклеотидные полиморфизмы в открытом доступе: насколько они полезны? Nat. Genet. 27 , 371–372 (2001).
CAS PubMed PubMed Central Google Scholar
Патил Н. и др.Блоки ограниченного разнообразия гаплотипов, выявленные сканированием с высоким разрешением хромосомы 21 человека. Science 294 , 1719–1723 (2001).
CAS Google Scholar
Коллинз, Ф.С., Гайер, М.С. И Чакраварти, А. Вариации на тему: каталогизация вариаций последовательности ДНК человека. Наука 278 , 1580–1581 (1997).
CAS PubMed PubMed Central Google Scholar
Габриэль С.B. et al. Структура гаплотипических блоков в геноме человека. Наука 296 , 2225–2229 (2002).
CAS PubMed PubMed Central Google Scholar
Дейли, М.Дж. и др. Структура гаплотипов высокого разрешения в геноме человека. Nat. Genet. 29 , 229–232 (2001).
CAS Google Scholar
Джеффрис, А.J., Kauppi, L. & Neumann, R. Интенсивно пунктированная мейотическая рекомбинация в области класса II главного комплекса гистосовместимости. Nat. Genet. 29 , 217–222 (2001).
CAS PubMed Google Scholar
Cargill, M. et al. Характеристика однонуклеотидных полиморфизмов в кодирующих областях генов человека. Nat. Genet. 22 , 231–238 (1999).
CAS PubMed PubMed Central Google Scholar
Галушка, м.K. et al. Паттерны однонуклеотидных полиморфизмов в генах-кандидатах на гомеостаз кровяного давления. Nat. Genet. 22 , 239–247 (1999).
CAS PubMed PubMed Central Google Scholar
Weiss, K.M. И Тервиллигер, J.D. Сколько болезней необходимо для картирования гена с помощью SNP? Nat. Genet. 26 , 151–157 (2000).
CAS PubMed Google Scholar
Райт, А.Ф. и Хасти, Н.Д. Сложные генетические заболевания: полемика по поводу кода Креза. Genome Biol. 2 , КОММЕНТАРИЙ 2007 (2001).
Altmuller, J. et al. Полногеномное сканирование сложных заболеваний человека: трудно найти истинную связь. г. J. Hum. Genet. 69 , 936–950 (2001).
CAS PubMed PubMed Central Google Scholar
Glatt, C.E. et al. Скрининг большого эталонного образца для выявления вариантов последовательностей с очень низкой частотой: сравнение двух генов. Nat. Genet. 27 , 435–438 (2001).
CAS PubMed Google Scholar
Дин М. и др. Типирование полиморфных примесей в этнических популяциях человека. г. J. Hum. Genet. 55 , 788–808 (1994).
CAS PubMed PubMed Central Google Scholar
Calafell, F. et al. Эволюция полиморфизма коротких тандемных повторов у человека. евро. J. Hum. Genet. 6 , 38–49 (1998).
CAS Google Scholar
Osier, M.V. и другие. Глобальный взгляд на генетические вариации в генах ADH обнаруживает необычные паттерны неравновесия по сцеплению и разнообразия. г. J. Hum. Genet. 71 , 84–99 (2002).
CAS PubMed PubMed Central Google Scholar
Мюллер-Мыхсок, Б.& Абель, Л. Генетический анализ сложных заболеваний. Наука 275 , 1328–1329 (1997).
CAS PubMed Google Scholar
Risch, N. & Teng, J. Относительная сила семейных планов и схем случай-контроль для исследований неравновесия сцепления сложных заболеваний человека I. Объединение ДНК. Genome Res. 8 , 1273–1288 (1998).
CAS PubMed Google Scholar
Хиршхорн, Дж.Н., Ломюллер, К., Бирн, Э. и Хиршхорн, К. Всесторонний обзор исследований генетических ассоциаций. Genet. Med. 4 , 45–61 (2002).
CAS PubMed PubMed Central Google Scholar
Сидоу, А. Последовательность сначала, задавайте вопросы позже. Cell 111 , 13–16 (2002).
CAS PubMed Google Scholar
Апарисио, С.и другие. Полногеномная сборка и анализ генома Fugu rubripes . Наука 297 , 1301–1310 (2002).
CAS PubMed PubMed Central Google Scholar
Базы данных генотипа-фенотипа человека: цели, проблемы и возможности
Джонстон, Дж. Дж. И Бизекер, Л. Г. Базы данных геномных вариаций и фенотипов: существующие ресурсы и будущие потребности. Hum.Мол. Genet. 22 , R27 – R31 (2013).
CAS PubMed PubMed Central Google Scholar
Рем, Х. Л. Секвенирование, ориентированное на заболевание: краеугольный камень в клинике. Nat. Преподобный Жене. 14 , 295–300 (2013).
CAS PubMed PubMed Central Google Scholar
Yang, Y. et al. Клиническое секвенирование всего экзома для диагностики менделевских расстройств. N. Engl. J. Med. 369 , 1502–1511 (2013).
CAS Статья PubMed PubMed Central Google Scholar
Zemojtel, T. et al. Эффективная диагностика генетического заболевания путем компьютерного анализа фенотипа связанного с заболеванием генома. Sci. Пер. Med. 6 , 252ra123 (2014).
PubMed PubMed Central Google Scholar
Сондерс, К.J. et al. Быстрое полногеномное секвенирование для диагностики генетических заболеваний в отделениях интенсивной терапии новорожденных. Sci. Пер. Med. 4 , 154ра135 (2012).
PubMed PubMed Central Google Scholar
Купер, Г. М. и Шендур, Дж. Иглы в стопках игл: поиск причинно-следственных вариантов заболевания в большом количестве геномных данных. Nat. Преподобный Жене. 12 , 628–640 (2011).
CAS PubMed Google Scholar
Ли, М.X. et al. Прогнозирование несинонимичных однонуклеотидных вариантов, вызывающих менделевскую болезнь, в исследованиях секвенирования экзома. PLoS Genet. 9 , e1003143 (2013).
CAS PubMed PubMed Central Google Scholar
Pelak, K. et al. Характеристика двадцати секвенированных геномов человека. PLoS Genet. 6 , e1001111 (2010).
PubMed PubMed Central Google Scholar
МакАртур, Д.G. et al. Систематический обзор вариантов потери функции в генах, кодирующих белки человека. Наука 335 , 823–828 (2012).
CAS PubMed PubMed Central Google Scholar
Horaitis, O. et al. База данных конкретных локусов. Nat. Genet. 39 , 425 (2007).
CAS PubMed Google Scholar
Патринос, Г.P. et al. Страновые узлы проекта Human Variome: документирование генетической информации внутри страны. Hum. Мутат. 33 , 1513–1519 (2012).
CAS PubMed Google Scholar
Fokkema, I. F. et al. LOVD v.2.0: новое поколение баз данных вариантов генов. Hum. Мутат. 32 , 557–563 (2011).
CAS PubMed Google Scholar
Бероуд, К., Collod-Beroud, G., Boileau, C., Soussi, T. и Junien, C. UMD (универсальная база данных мутаций): универсальное программное обеспечение для создания и анализа баз данных, специфичных для локусов. Hum. Мутат. 15 , 86–94 (2000). Ссылки 12 и 13 описывают две наиболее часто используемые программные платформы для создания LSDB и для аннотирования, анализа и отображения вариаций ДНК в генах.
CAS PubMed Google Scholar
Польви, А.и другие. Обновленная база данных финского наследия болезней (FinDis) — база данных для генов, мутировавших в финском наследии болезней, перенесенная в эру секвенирования следующего поколения. Hum. Мутат. 34 , 1458–1466 (2013).
PubMed Google Scholar
Dalgleish, R. et al. Ссылка на локус Геномные последовательности: улучшенная основа для описания вариантов ДНК человека. Genome Med. 2 , 24 (2010).
PubMed PubMed Central Google Scholar
MacArthur, J. A. et al. Геномный эталон локуса: эталонные последовательности для сообщения о клинически значимых вариантах последовательностей. Nucleic Acids Res. 42 , D873 – D878 (2014).
CAS PubMed Google Scholar
Подагра, A. M. et al. Анализ опубликованных вариантов последовательности гена PKD1 . Nat. Genet. 39 , 427–428 (2007).
CAS PubMed Google Scholar
Bell, C.J. et al. Тестирование носителей при тяжелых детских рецессивных заболеваниях с помощью секвенирования следующего поколения. Sci. Пер. Med. 3 , 65ра4 (2011).
CAS PubMed PubMed Central Google Scholar
Chen, S. N. et al.Молекулярно-генетические и функциональные исследования человека идентифицируют TRIM63 , кодирующий мышечный белок 1 пальца RING, как новый ген гипертрофической кардиомиопатии человека. Circ. Res. 111 , 907–919 (2012).
CAS PubMed PubMed Central Google Scholar
Ploski, R. et al. Вызывает ли p.Q247X в TRIM63 гипертрофическую кардиомиопатию человека? Circ. Res. 114 , e2 – e5 (2014).
CAS PubMed Google Scholar
Witt, C.C. et al. Кооперативный контроль поперечно-полосатой мышечной массы и метаболизма с помощью MuRF1 и MuRF2. EMBO J. 27 , 350–360 (2008).
CAS PubMed Google Scholar
Plon, S.E. et al. Классификация вариантов последовательности и составление отчетов: рекомендации по улучшению интерпретации результатов генетических тестов на предрасположенность к раку. Hum. Мутат. 29 , 1282–1291 (2008).
CAS PubMed PubMed Central Google Scholar
Соснай П. Р. и др. Определение предрасположенности к заболеванию вариантов гена регулятора трансмембранной проводимости при муковисцидозе. Nat. Genet. 45 , 1160–1167 (2013).
CAS PubMed PubMed Central Google Scholar
den Dunnen, J., Каттинг, Г. Р. и Паалман, М. Х. Обязательное представление варианта — наш опыт. Hum. Мутат. 33 , 1 (2012).
Google Scholar
Терри, С.Ф. Организации по защите интересов болезней катализируют трансляционные исследования. Фронт. Genet. 4 , 101 (2013).
PubMed PubMed Central Google Scholar
Уикс, П.и другие. Обмен данными о здоровье для улучшения результатов на PatientsLikeMe. J. Med. Интернет. Res. 12 , e19 (2010).
PubMed PubMed Central Google Scholar
Киркпатрик, Б. Э. и др. GenomeConnect: поиск партнеров, клинических лабораторий и исследователей для улучшения геномных знаний. Hum. Мутат. 36 , 974–978 (2015).
PubMed PubMed Central Google Scholar
Макаллистер, М.& Dearing, A. Пациент сообщил об исходах и расширении прав и возможностей пациентов в службах клинической генетики. Clin. Genet. 88 , 114–121 (2015).
CAS PubMed Google Scholar
Редакция журнала «Ланцет». Расширение прав и возможностей пациентов — кто кого уполномочивает? Ланцет 379 , 1677 (2012).
Abecasis, G. R. et al. Карта вариаций генома человека по результатам популяционного секвенирования. Природа 467 , 1061–1073 (2010).
Артикул PubMed Google Scholar
Tennessen, J. A. et al. Эволюция и функциональное влияние редких вариантов кодирования из глубокого секвенирования экзомов человека. Наука 337 , 64–69 (2012).
CAS PubMed PubMed Central Google Scholar
Болл, М. П.и другие. Общедоступный ресурс, облегчающий клиническое использование геномов. Proc. Natl Acad. Sci. США 109 , 11920–11927 (2012).
CAS PubMed Google Scholar
Zhang, J. et al. Портал данных Международного консорциума генома рака — универсальный магазин данных по геномике рака. База данных (Оксфорд) 2011 , bar026 (2011). Портал данных ICGC предоставляет инструменты для визуализации, запроса и загрузки огромного количества данных из ICGC с инновационным подходом к объединению данных и аннотаций между многочисленными участвующими центрами.
Google Scholar
Weinstein, J. N. et al. Проект «Атлас генома рака» Пан-раковый аналитический проект. Nat. Genet. 45 , 1113–1120 (2013).
PubMed PubMed Central Google Scholar
Петровски, С. и Гольдштейн, Д. Б. Феномика и интерпретация личных геномов. Sci. Пер. Med. 6 , 254fs35 (2014).
PubMed Google Scholar
Доршнер, М. О. и др. Подходящие к действию патогенные случайные находки в экзомах 1000 участников. г. J. Hum. Genet. 93 , 631–640 (2013). Этот отчет убедительно демонстрирует, что часто недостаточно доказательств патогенности многих вариантов, описанных в базах данных или в медицинской литературе.
CAS PubMed PubMed Central Google Scholar
Димент, Д.A. et al. Секвенирование всего экзома расширяет фенотипический спектр редкой детской эпилепсии: ретроспективное исследование. Clin. Genet. 88 , 34–40 (2015).
CAS PubMed Google Scholar
Firth, H. V. et al. ДЕЦИФЕР: База данных хромосомного дисбаланса и фенотипа у людей с использованием ресурсов ансамбля. г. J. Hum. Genet. 84 , 524–533 (2009). DECIPHER — это крупнейшая общедоступная база данных генотипических и фенотипических данных в основном недиагностированных пациентов с редкими заболеваниями.DECIPHER имеет большой набор инструментов для облегчения интерпретации возможных вариантов.
CAS PubMed PubMed Central Google Scholar
Гонсалес, М.А. и др. Приложение GEnomes Management (GEM.app): новый программный инструмент для крупномасштабного совместного анализа генома. Hum. Мутат. 34 , 842–846 (2013).
CAS PubMed PubMed Central Google Scholar
Денни, Дж.C. et al. Систематическое сравнение исследования ассоциации по всему феномену данных электронных медицинских карт и данных исследования ассоциации по всему геному. Nat. Biotechnol. 31 , 1102–1110 (2013).
CAS PubMed PubMed Central Google Scholar
Schaefer, C. & RPGEH GO Project Collaboration. Программа исследований Kaiser Permanente по генам, окружающей среде и здоровью: ресурс по генетической эпидемиологии здоровья и старения взрослых. Clin. Med. Res. 9 , 177–178 (2011).
PubMed Central Google Scholar
Lappalainen, I. et al. Европейский архив геномных данных о людях дал согласие на проведение биомедицинских исследований. Nat. Genet. 47 , 692–695 (2015).
CAS PubMed PubMed Central Google Scholar
Tryka, K. A. et al.База данных генотипов и фенотипов NCBI: dbGaP. Nucleic Acids Res. 42 , D975 – D979 (2014).
CAS PubMed Google Scholar
Кохан И.С. Использование электронных медицинских карт для открытия в геномике болезней. Nat. Преподобный Жене. 12 , 417–428 (2011).
CAS PubMed Google Scholar
Мерфи, С.N. et al. Обслуживание предприятия и не только с помощью информатики для интеграции биологии и прикроватной работы (i2b2). J. Am. Med. Поставить в известность. Доц. 17 , 124–130 (2010).
PubMed PubMed Central Google Scholar
Adamusiak, T. et al. Observ-OM и Observ-TAB: универсальные синтаксические решения для интеграции, поиска и обмена информацией о фенотипах и генотипах. Hum. Мутат. 33 , 867–873 (2012).
PubMed Google Scholar
Бек, Т., Гастингс, Р.К., Голлапуди, С., Фри, Р.С. и Брукс, А.Дж. GWAS Central: всеобъемлющий ресурс для сравнения и анализа полногеномных ассоциативных исследований. евро. J. Hum. Genet. 22 , 949–952 (2014). Самый крупный общедоступный сборник сводных результатов исследований генетических ассоциаций. Вместе со ссылками 50 и 111–113 это обеспечивает альтернативные способы поиска и визуализации данных GWAS.
PubMed Google Scholar
Gaye, A. et al. DataSHIELD: анализ данных, а не данных для анализа. Внутр. J. Epidemiol. 43 , 1929–1944 (2014).
PubMed PubMed Central Google Scholar
Karr, A. et al. Безопасный анализ распределенных баз данных с сохранением конфиденциальности. Technometrics 49 , 335–345 (2007).
Google Scholar
Кариазо, М. и Леннон, Г. SNPedia: вики-сайт, поддерживающий аннотацию, интерпретацию и анализ личного генома. Nucleic Acids Res. 40 , D1308 – D1312 (2012).
CAS PubMed Google Scholar
Rappaport, N. et al. MalaCards: интегрированный сборник болезней и их аннотации. База данных (Оксфорд) 2013 , bat018 (2013).
Google Scholar
Лопес П., Далглиш Р. и Оливейра Дж. Л. WAVe: веб-анализ вариома. Hum. Мутат. 32 , 729–734 (2011).
CAS PubMed Google Scholar
Glusman, G., Caballero, J., Mauldin, D. E., Hood, L. & Roach, J. C. Kaviar: доступная система для тестирования новизны SNV. Биоинформатика 27 , 3216–3217 (2011).
CAS PubMed PubMed Central Google Scholar
Филиппакис, А.А. и др. Биржа свах: платформа для открытия генов редких болезней. Hum. Мутат. 36 , 915–921 (2015).
PubMed PubMed Central Google Scholar
Manolio, T. A. et al. Глобальное внедрение геномной медицины: мы не одиноки. Sci. Пер. Med. 7 , 290ps13 (2015).
PubMed PubMed Central Google Scholar
Хайден, Э. К. Генетики настаивают на глобальном обмене данными. Природа 498 , 16–17 (2013). Отчет об основании GA4GH.
PubMed Google Scholar
Gottlieb, M. M. et al. GeneYenta: инструмент для сопоставления случаев редкого заболевания на основе фенотипа, основанный на алгоритмах онлайн-датирования для ускорения интерпретации экзома. Hum. Мутат. 36 , 432–438 (2015).
PubMed Google Scholar
Lancaster, O. et al. Cafe Variome: универсальное программное обеспечение для обнаружения данных генотип-фенотип в контексте ограниченного или открытого доступа. Hum. Мутат. 36 , 957–964 (2015).
PubMed Google Scholar
Wellcome Trust. Повышение доступности данных исследований общественного здравоохранения и эпидемиологии. Wellcome Trust [онлайн], (2014).
Цифровой кулинарный центр (DCC). Jisc Research Data Registry and Discovery Service DCC http://www.dcc.ac.uk/projects/research-data-registry-pilot (2014).
Коттон, Р. Г. и др. Проект человеческого вариома. Наука 322 , 861–862 (2008).
CAS PubMed PubMed Central Google Scholar
Рем, Х.L. et al. ClinGen — Ресурс клинического генома. N. Engl. J. Med. 372 , 2235–2242 (2015). ClinGen — это ресурс, финансируемый Национальным институтом здравоохранения, который создает авторитетную базу знаний, которая продвигает основанные на фактах клинические аннотации и интерпретацию геномных вариантов. ClinVar (ссылка 61), активный партнер проекта ClinGen, представляет собой свободно доступный общедоступный архив отчетов о взаимосвязях между человеческими вариациями и фенотипами с подтверждающими доказательствами.
CAS PubMed PubMed Central Google Scholar
Landrum, M. J. et al. ClinVar: общедоступный архив взаимосвязей между вариациями последовательностей и фенотипом человека. Nucleic Acids Res. 42 , D980 – D985 (2014).
CAS PubMed Google Scholar
Beaulieu, C. L. et al. Консорциум FORGE Canada: результаты двухлетнего национального проекта по открытию генов редких заболеваний. г. J. Hum. Genet. 94 , 809–817 (2014).
CAS PubMed PubMed Central Google Scholar
Буске, О. Дж. PhenomeCentral: портал для фенотипического и генотипического сопоставления пациентов с редкими генетическими заболеваниями. Hum. Мутат. 36 , 931–940 (2015).
PubMed PubMed Central Google Scholar
Томпсон, Р.и другие. RD-Connect: интегрированная платформа, объединяющая базы данных, реестры, биобанки и клиническую биоинформатику для исследования редких заболеваний. J. Gen. Intern. Med. 29 , S780 – S787 (2014).
PubMed Google Scholar
Конли, Дж. М., Кук-Диган, Р. и Ласаро-Муньос, Г. Мириады после Мириады : дилемма частных данных. N. C. J. Law Technol. 15 , 597–637 (2014).
PubMed PubMed Central Google Scholar
Риггс, Э. Р., Джексон, Л., Миллер, Д. Т. и Ван Вурен, С. Фенотипическая информация в базах данных геномных вариантов улучшает клиническую помощь и исследования: опыт Консорциума международных стандартов для цитогеномных массивов. Hum. Мутат. 33 , 787–796 (2012).
PubMed PubMed Central Google Scholar
Хадсон, Т.J. et al. Международная сеть проектов генома рака. Природа 464 , 993–998 (2010).
CAS PubMed Google Scholar
Banda, Y. et al. Характеристика расы / этнической принадлежности и генетического происхождения для 100000 субъектов в когорте генетических эпидемиологических исследований здоровья и старения взрослых (GERA). Генетика 200 , 1285–1295 (2015).
PubMed PubMed Central Google Scholar
Кнопки, Б.M. Структура ответственного обмена геномными данными и данными, связанными со здоровьем. HUGO J. 8 , 3 (2014). Отчет об этических рамках для ответственного обмена данными, разработанный совместно с широким спектром биоэтических, геномных и клинических сообществ под эгидой GA4GH.
PubMed PubMed Central Google Scholar
Маскальцони, Д. и др. Международная хартия принципов обмена биопрепаратами и данными. евро. J. Hum. Genet. 23 , 721–728 (2015).
PubMed Google Scholar
Rath, A. et al. Представление редких заболеваний в информационных системах здравоохранения: подход Orphanet для обслуживания широкого круга конечных пользователей. Hum. Мутат. 33 , 803–808 (2012). Orphanet — это портал для редких болезней и орфанных лекарств, который содержит перечень редких заболеваний и систему классификации, которая служит моделью для обновления международной терминологии, такой как Международная классификация болезней.
PubMed Google Scholar
Köhler, S. et al. Проект «Онтология фенотипа человека»: связь молекулярной биологии и болезней посредством фенотипических данных. Nucleic Acids Res. 42 , D966 – D974 (2014). Отчет об онтологии человеческого фенотипа, широко используемом стандарте для аннотирования и анализа фенотипических аномалий в диагностических и трансляционных исследованиях.
PubMed Google Scholar
Варга, Э.А. и Молл, С. Страницы пациентов кардиологии. Мутация протромбина 20210 (мутация фактора II). Тираж 110 , e15 – e18 (2004 г.).
PubMed Google Scholar
Beaudet, A. L. & Tsui, L. C. Предлагаемая номенклатура для обозначения мутаций. Hum. Мутат. 2 , 245–248 (1993).
CAS PubMed Google Scholar
ден Даннен, Дж.T. & Antonarakis, S.E. Номенклатура для описания вариаций последовательностей человека. Hum. Genet. 109 , 121–124 (2001). Начальное описание номенклатурного стандарта Общества вариаций генома человека для наименования вариантов последовательностей.
CAS PubMed Google Scholar
Ташнер, П. Э. и ден Даннен, Дж. Т. Описание структурных изменений путем расширения номенклатуры вариаций последовательности HGVS. Hum. Мутат. 32 , 507–511 (2011).
CAS PubMed Google Scholar
Ларос, Дж. Ф., Блавье, А., ден Даннен, Дж. Т. и Ташнер, П. Э. Формализованное описание стандартной номенклатуры вариантов человека в расширенной форме Бэкуса-Наура. BMC Bioinformatics 12 , S5 (2011).
PubMed PubMed Central Google Scholar
Уайлдман, М., van Ophuizen, E., den Dunnen, J. T. & Taschner, P. E. Улучшение описаний вариантов последовательностей в базах данных по мутациям и литературе с использованием программы проверки номенклатуры вариантов последовательностей Mutalyzer. Hum. Мутат. 29 , 6–13 (2008).
CAS PubMed Google Scholar
Hart, R.K. et al. Пакет Python для анализа, проверки, сопоставления и форматирования вариантов последовательностей с использованием номенклатуры HGVS. Биоинформатика 31 , 268–270 (2015).
CAS PubMed Google Scholar
Кок, П. Дж., Филдс, К. Дж., Гото, Н., Хойер, М. Л. и Райс, П. М. Формат файла Sanger FASTQ для последовательностей с оценками качества и вариантов Solexa / Illumina FASTQ. Nucleic Acids Res. 38 , 1767–1771 (2010).
CAS PubMed Google Scholar
Li, H. et al. Формат Sequence Alignment / Map и SAMtools. Биоинформатика 25 , 2078–2079 (2009).
PubMed PubMed Central Google Scholar
Danecek, P. et al. Вариант формата вызова и VCFtools. Биоинформатика 27 , 2156–2158 (2011).
CAS PubMed PubMed Central Google Scholar
Бирн, М. и др. Платформа VarioML для комплексного представления данных о вариациях и обмена ими BMC Bioinformatics 13 , 254 (2012).
PubMed PubMed Central Google Scholar
Вихинен, М. Вариационная онтология для аннотации вариационных эффектов и механизмов. Genome Res. 24 , 356–364 (2014).
CAS PubMed PubMed Central Google Scholar
Kibbe, W. A. et al. Обновление Disease Ontology 2015: расширенная и обновленная база данных по заболеваниям человека для увязки биомедицинских знаний с данными о болезнях. Nucleic Acids Res. 43 , D1071 – D1078 (2015).
CAS PubMed Google Scholar
Groza, T. et al. Онтология фенотипа человека: семантическое объединение общих и редких заболеваний. г. J. Hum. Genet. 97 , 111–124 (2015).
CAS PubMed PubMed Central Google Scholar
Райт, К. Ф.и другие. Генетическая диагностика нарушений развития в исследовании DDD: масштабируемый анализ данных полногеномного исследования. Ланцет 385 , 1305–1314 (2015).
PubMed PubMed Central Google Scholar
Sifrim, A. et al. eXtasy: приоритезация вариантов путем слияния геномных данных. Nat. Методы 10 , 1083–1084 (2013).
CAS PubMed Google Scholar
Робинсон, П.N. et al. Улучшенная приоритезация генов болезней экзомами посредством межвидового сравнения фенотипов. Genome Res. 24 , 340–348 (2014).
CAS PubMed PubMed Central Google Scholar
Синглтон, М. В. и др. Phevor объединяет несколько биомедицинских онтологий для точной идентификации вызывающих болезнь аллелей у отдельных людей и небольших нуклеарных семей. г. J. Hum. Genet. 94 , 599–610 (2014).
CAS PubMed PubMed Central Google Scholar
Джавед А., Агравал С. и Нг, П. С. Фен-Ген: сочетание фенотипа и генотипа для анализа редких заболеваний. Nat. Методы 11 , 935–937 (2014).
CAS PubMed Google Scholar
Westbury, S. K. et al. Аннотации онтологии фенотипа человека и кластерный анализ для выявления генетических дефектов в 707 случаях с необъяснимыми кровотечениями и нарушениями тромбоцитов. Genome Med. 7 , 36 (2015).
PubMed PubMed Central Google Scholar
Адам Д. Психическое здоровье: в спектре. Природа 496 , 416–418 (2013).
CAS PubMed Google Scholar
Stenson, P. D. et al. База данных мутаций генов человека: к всеобъемлющей центральной базе данных мутаций. Дж.Med. Genet. 45 , 124–126 (2008).
CAS PubMed Google Scholar
Абель, О., Пауэлл, Дж. Ф., Андерсен, П. М. и Аль-Чалаби, А. ALSoD: удобный онлайн-инструмент биоинформатики для генетики бокового амиотрофического склероза. Hum. Мутат. 33 , 1345–1351 (2012).
CAS PubMed Google Scholar
Чандрасекхараппа, С.C. et al. Технологии массового параллельного секвенирования, aCGH и RNA-seq обеспечивают комплексную молекулярную диагностику анемии Фанкони. Кровь 121 , e138 – e148 (2013).
CAS PubMed PubMed Central Google Scholar
Dalgleish, R. База данных мутаций коллагена человека типа I. Nucleic Acids Res. 25 , 181–187 (1997).
CAS PubMed PubMed Central Google Scholar
Пийрила, Х., Валиахо, Дж. И Вихинен, М. Базы данных мутаций иммунодефицита (IDbases). Hum. Мутат. 27 , 1200–1208 (2006).
CAS PubMed Google Scholar
Ruiz-Pesini, E. et al. Улучшенный MITOMAP с глобальной мутационной филогенией мтДНК. Nucleic Acids Res. 35 , D823 – D828 (2007).
CAS PubMed Google Scholar
Пападопулос, П.и другие. Разработки всемирной базы данных FINDbase для определения частот аллелей клинически значимых геномных вариаций. Nucleic Acids Res. 42 , D1020 – D1026 (2014).
CAS PubMed Google Scholar
Брагин Э. и др. DECIPHER: база данных для интерпретации вероятной патогенной последовательности, связанной с фенотипом, и вариации числа копий. Nucleic Acids Res. 42 , D993 – D1000 (2014).
CAS PubMed Google Scholar
Hamosh, A. et al. PhenoDB: новый веб-инструмент для сбора, хранения и анализа фенотипических признаков. Hum. Мутат. 34 , 566–571 (2013).
CAS PubMed PubMed Central Google Scholar
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: инструмент сопоставления для подключения исследователей, заинтересованных в одном и том же гене. Hum. Мутат. 36 , 928–930 (2015).
PubMed PubMed Central Google Scholar
Амбергер, Дж., Боккини, К. и Хамош, А. Новое лицо и новые проблемы для онлайн-менделевского наследования в человеке (OMIM ® ). Hum. Мутат. 32 , 564–567 (2011). OMIM — одна из старейших и наиболее важных баз знаний в области медицины человека, восходящая к работе, начатой в начале 1960-х годов Виктором МакКусиком.Помимо 12 книжных изданий книги Mendelian Inheritance in Man (MIM) в период с 1966 по 1998 год, с 1987 года доступна онлайн-версия (OMIM).
PubMed Google Scholar
Mungall, C.J. et al. Использование баз данных модельных организмов и болезней для поддержки поиска генов болезней человека. Hum. Мутат. 36 , 979–984 (2015).
PubMed PubMed Central Google Scholar
Уилкс, К.и другие. Центр геномики рака (CGHub): преодоление рака с помощью огромного количества данных. База данных (Оксфорд) 2014 , bau093 (2014).
Google Scholar
Forbes, S.A. et al. COSMIC: поиск полных геномов рака в Каталоге соматических мутаций рака. Nucleic Acids Res. 39 , D945 – D950 (2011).
CAS PubMed Google Scholar
Cheng, W.C. et al. DriverDB: база данных секвенирования экзома для идентификации генов-драйверов рака. Nucleic Acids Res. 42 , D1048 – D1054 (2014).
CAS PubMed Google Scholar
Welter, D. et al. Каталог NHGRI GWAS, кураторский ресурс ассоциаций SNP-признаков. Nucleic Acids Res. 42 , D1001 – D1006 (2014).
CAS PubMed Google Scholar
Ли, М.J. et al. GWASdb: база данных генетических вариантов человека, выявленных в результате полногеномных ассоциативных исследований. Nucleic Acids Res. 40 , D1047 – D1054 (2012).
CAS PubMed Google Scholar
Коике, А., Нисида, Н., Иноуэ, И., Цудзи, С. и Токунага, К. Общегеномная ассоциативная база данных, разработанная в рамках японского проекта интегрированной базы данных. J. Hum. Genet. 54 , 543–546 (2009).
PubMed Google Scholar
Whirl-Carrillo, M. et al. Знания фармакогеномики для персонализированной медицины. Clin. Pharmacol. Ther. 92 , 414–417 (2012).
CAS PubMed PubMed Central Google Scholar
Кэри, Дж. К., Аллансон, Дж. Э., Хеннекам, Р. К. и Бизекер, Л. Г. Стандартная терминология для фенотипических вариаций: элементы проекта морфологии, его текущий прогресс и будущие направления. Hum. Мутат. 33 , 781–786 (2012).
PubMed Google Scholar
Группы крови ABO: прогнозирование группы крови ваших детейВступлениеУчебное пособие по генетике человека с упражнениями по решению проблем, касающихся наследования аллелей группы крови ABO, привело к постоянному потоку запросов в Проект по биологии от матерей, бабушек и детей, спрашивающих о возможной группе крови отца данного ребенка.Вот типичный запрос: Я читал вашу информацию о наследовании групп крови и очень запутался! Я пытаюсь выяснить, какая группа крови отец моего сына мог, так как мы с сыном оба типа А +. Также, мой брат — тип 0, а моя мама — пятерка. Мы не можем найти ничего, что объясняет, как это может быть. Не могли бы вы помочь ???. Человеческие маркеры ABO: аллели A, B и O Группа крови человека определяется кодоминантными аллелями.Аллель — это одна из нескольких различных форм генетической информации, которая присутствует в нашей ДНК в определенном месте на определенной хромосоме. Есть три разных аллеля группы крови человека, известные как I A , I B и i. Для простоты мы можем назвать эти аллели A (для I A ), B (для I B ) и O (для i). У каждого из нас есть два аллеля группы крови ABO, потому что каждый из нас наследует один аллель группы крови от нашей биологической матери и один от нашего биологического отца.Описание пары аллелей в нашей ДНК называется генотипом. Поскольку существует три разных аллеля, всего в генетическом локусе ABO человека имеется шесть различных генотипов. Возможные генотипы — AA, AO, BB, BO, AB и OO. Как группы крови связаны с шестью генотипами?Анализ крови используется, чтобы определить, присутствуют ли в образце крови характеристики A и / или B. Невозможно определить точный генотип по результатам анализа крови типа A или типа B.Если у кого-то группа крови A, у него должна быть по крайней мере одна копия аллеля A, но у них может быть две копии. Их генотип — AA или AO. Точно так же человек с группой крови B может иметь генотип BB или BO. Анализ крови типа AB или O более информативен. Человек с группой крови AB должен иметь аллели как A, так и B. Генотип должен быть AB. У человека с группой крови O нет ни аллелей A, ни B. Генотип должен быть OO. Как наши дети наследуют аллели ABO?Каждый биологический родитель жертвует своему ребенку один из двух аллелей ABO.Мать с группой крови O может передать аллель O только своему сыну или дочери. Отец с группой крови AB может передать аллель A или B своему сыну или дочери. У этой пары могут быть дети либо группы крови A (O от матери и A от отца), либо группы крови B (O от матери и B от отца). Поскольку существует 4 различных группы крови матери и 4 разные группы крови отца, существует 16 различных комбинаций, которые следует учитывать при прогнозировании группы крови детей.В таблицах ниже показаны все 16 возможных комбинаций. Если вы знаете группу крови матери и отца, можно определить возможные группы крови для их детей. А как насчет резус-фактора? Может ли отец с группой крови A + иметь ребенка с группой крови A-?Генетическая информация о резус-факторе также передается по наследству от наших родителей, но наследуется независимо от аллелей группы крови ABO. Существует 2 разных аллеля резус-фактора, известных как Rh + и Rh-.У кого-то «Rh-положительный» или «Rh +» есть по крайней мере один аллель Rh +, но может быть и два. Их генотип мог быть либо Rh + / Rh +, либо Rh + / Rh-. Тот, кто Rh- имеет генотип Rh- / Rh-. Как и в случае с аллелями ABO, каждый биологический родитель передает своему ребенку один из двух своих аллелей Rh. Мать-резус-фактор может передать резус-аллель только своему сыну или дочери. Отец с Rh + может передать Rh + или Rh- аллель своему сыну или дочери. У этой пары могли быть Rh + дети (Rh- от матери и Rh + от отца) или Rh- дети (Rh- от матери и Rh- от отца). Отвечая на вопрос матери из Альберты, КанадаРассматриваемая мать — группа крови А +. Ее генотип в местоположении ABO — AA или AO. Ее генотип Rh — либо Rh + / Rh +, либо Rh + / Rh-. Информация о том, что у бабушки по материнской линии также группа крови A +, а у брата — группа крови O, говорит нам, что бабушка по материнской линии ребенка имеет генотип AO, поскольку она принадлежит к типу A, но передала аллель O одному из своих детей. Мать хочет знать потенциальные группы крови отца ее сына.Сын группы крови А +. К сожалению, в этом конкретном случае мать не может отличить потенциальных отцов только по группе крови. Обратите внимание на таблицу, что эта мать могла родить ребенка с кровью типа A от отца любой из четырех возможных групп крови: типа A, типа AB, типа B или типа O. Аналогичным образом, отцом ребенка может быть любой Rh + или Rh-. Из этого обсуждения должно быть очевидно, что группа крови — не очень хороший тест на отцовство.В некоторых случаях может быть получена однозначная информация, например, мужчина типа AB не может стать отцом ребенка типа O. Однако в большинстве случаев результаты сомнительны. Если определение отцовства ребенка важно, в настоящее время доступны очень чувствительные ДНК-тесты, которые могут установить отцовство с достоверностью, превышающей 99,99%, или исключить кого-то из биологического отца с абсолютной уверенностью. В другом месте проекта «Биология» есть упражнение по отслеживанию наследования ДНК-маркеров в исследовании отцовства. Справочные таблицы по группе крови
Ричард Б.Халлик Университет Аризоны 26 авг.1997 г. [email protected] http://www.blc.arizona.edu/ |
Генотип против фенотипа: примеры и определения
Любой организм — это побочный продукт своей генетической структуры и окружающей среды. Чтобы понять это в деталях, мы должны сначала оценить некоторые основные генетические термины и концепции.Здесь мы даем определения терминов генотип и фенотип , обсуждаем их взаимосвязь и рассматриваем, почему и как мы могли бы их изучить.
Что такое определение генотипа?
В биологии ген — это часть ДНК, которая кодирует признак. Точное расположение нуклеотидов (каждый состоит из фосфатной группы, сахара и основания) в гене может различаться в разных копиях одного и того же гена. Следовательно, ген может существовать в разных формах у разных организмов.Эти разные формы известны как аллели. Точное фиксированное положение на хромосоме, содержащее конкретный ген, называется локусом.
Диплоидный организм наследует либо две копии одного и того же аллеля, либо одну копию двух разных аллелей от своих родителей. Если человек наследует два идентичных аллеля, его генотип считается гомозиготным по этому локусу.
Однако, если они обладают двумя разными аллелями, их генотип считается гетерозиготным по этому локусу. Аллели одного и того же гена могут быть аутосомно-доминантными или рецессивными.Аутосомно-доминантный аллель всегда будет предпочтительно выражаться по сравнению с рецессивным аллелем.
Последующая комбинация аллелей, которыми обладает человек для определенного гена, — это его генотип .
Примеры генотиповРассмотрим классический пример — цвет глаз.
- Ген кодирует цвет глаз.
- В этом примере аллель либо коричневый, либо синий, причем один унаследован от матери, а другой — от отца.
- Коричневый аллель является доминантным (B), а синий аллель — рецессивным (b). Если ребенок наследует два разных аллеля (гетерозиготных), у него будут карие глаза. Чтобы у ребенка были голубые глаза, они должны быть гомозиготными по аллелю голубого глаза.
Рис. 1: Диаграмма наследования, в которой подробно показано, как индивид может унаследовать голубые или карие глаза в зависимости от аллелей, принадлежащих их родителям, причем аллель цвета карих глаз является доминантным, а аллель цвета голубых глаз — рецессивным.
Другие примеры генотипа включают:
- Цвет волос
- Рост
- Размер обуви
Сумма наблюдаемых характеристик организма — это их фенотип. Ключевое различие между фенотипом и генотипом состоит в том, что в то время как генотип унаследован от родителей организма, фенотип — нет.
Хотя фенотип влияет на генотип, генотип не равен фенотипу.На фенотип влияет генотип и факторы, включая:
- Эпигенетические модификации
- Факторы окружающей среды и образа жизни
Рисунок 2: Фламинго от природы имеют белый цвет, их вызывают только пигменты организмов, которые они едят чтобы стать ярко-розовым.
Примеры фенотипов
Факторы окружающей среды, которые могут влиять на фенотип, включают питание, температуру, влажность и стресс.Фламинго — классический пример того, как окружающая среда влияет на фенотип. Несмотря на то, что они известны своим ярко-розовым цветом, их естественный цвет — белый — розовый цвет вызван пигментами организмов в их рационе.
Второй пример — цвет кожи человека. Наши гены контролируют количество и тип меланина, который мы производим, однако воздействие ультрафиолетового света в солнечном климате вызывает потемнение существующего меланина и способствует усилению меланогенеза и, следовательно, более темной коже.
Генотип против фенотипа: наблюдениеНаблюдать за фенотипом просто — мы смотрим на внешние особенности и характеристики организма и делаем выводы о них.Однако наблюдение за генотипом немного сложнее.
Генотипирование — это процесс, при котором различия в генотипе человека анализируются с помощью биологических анализов. Полученные данные затем можно сравнить либо с последовательностью второго человека, либо с базой данных последовательностей.
Ранее генотипирование позволяло получить только частичные последовательности. Теперь, благодаря крупным технологическим достижениям последних лет, мы получили самое современное секвенирование всего генома.
Рисунок 3: Рабочий процесс, изображающий различные этапы секвенирования всего генома (WGS).
(WGS) позволяет получать целые последовательности. WGS — это эффективный процесс, который становится все более доступным и предполагает использование высокопроизводительных методов секвенирования, таких как секвенирование одной молекулы в реальном времени (SMRT) для идентификации исходной последовательности нуклеотидов, составляющих ДНК организма.
WGS — это не единственный способ анализа генома организма — доступны различные методы.
Почему важно изучать генотип и фенотип?Понимание взаимосвязи между генотипом и фенотипом может быть чрезвычайно полезным в различных областях исследований.
Особенно интересная область — фармакогеномика. Генетические вариации могут возникать в ферментах печени, необходимых для метаболизма лекарств, таких как CYP450. Следовательно, фенотип человека, то есть его способность метаболизировать конкретное лекарство, может варьироваться в зависимости от того, какой формой гена, кодирующего фермент, он обладает. Для фармацевтических компаний и врачей эти знания являются ключевыми для определения рекомендуемых дозировок лекарств для разных групп населения.
Тандемное использование методов генотипирования и фенотипирования оказывается лучше, чем использование только генотипических тестов.В сравнительном клиническом исследовании фармакогеномики подход мультиплексирования выявил большие различия в способности метаболизма лекарств, чем это было предсказано только генотипированием. Это имеет важное значение для персонализированной медицины и подчеркивает необходимость проявлять осторожность, полагаясь исключительно на генотипирование.
Как мы можем изучить взаимосвязь между генотипом и фенотипом?Используя животных моделей, таких как мыши, ученые могут генетически модифицировать организм так, чтобы он больше не экспрессировал определенный ген — известные как нокаутные мыши.Сравнивая фенотип этого животного с фенотипом дикого типа (то есть фенотипом, который существует, когда ген не был удален), мы можем изучить роль определенных генов в обеспечении определенных фенотипов.
Инициатива в области информатики генома мышей (MGI) собрала базу данных тысяч фенотипов, которые могут быть созданы и изучены, а также генов, которые должны быть выбиты для получения каждого конкретного фенотипа.
Таблица генотипов и фенотипов:| | Генотип | Фенотип | |||
| 9307 930 930 9309 отвечает за определенный признак | Наблюдаемые характеристики и признаки организма | ||||
| Характеризуется | Методы генотипирования, такие как WGS | Наблюдение за внешними характеристиками организма | 000 | Последовательности генов, которыми обладает организм | Генотип, ПЛЮС эпигенетика и факторы окружающей среды |
| Унаследовано? | Да | Нет | |||
| Пример | Гены, кодирующие цвет глаз | Человек с карими глазами |
Генетика человека — Проект по генетике и генетике человека и оценка риска
Гены — это фундаментальные единицы наследственности, а геном — это совокупность генов организма.Генотип — это уникальный набор всех генов отдельного организма. Генотип сложным образом управляет фенотипом, который представляет собой совокупность всех черт внешнего вида, функций и поведения организма. В настоящее время известно, что гены представляют собой последовательности дезоксирибонуклеиновой кислоты (ДНК), из которых транскрибируется рибонуклеиновая кислота (РНК). Транскрипты большинства, но не всех генов затем транслируются в белки, которые состоят из аминокислотных последовательностей и выполняют большинство функций клетки благодаря своей каталитической активности и взаимодействиям, происходящим в их специфических сайтах связывания.Следовательно, ген необходим для фенотипического признака, потому что он кодирует белок, участвующий в формировании признака.
Точно неизвестно, сколько генов содержит геном человека, но оценки варьируются от 61 000 до 140 000 (Dickson 1999; Dunham et al. 1999). Для сравнения, нематода Caenorhabditis elegans имеет 19 000 генов. У человека только 5% ДНК генома действительно кодирует белки. Остальные служат либо в качестве регуляторных последовательностей, которые определяют условия, при которых ген будет транскрибироваться, либо в качестве интронов (последовательности, которые транскрибируются, но не транслируются), либо в качестве спейсерной ДНК, функция которой пока неизвестна.Каждый ген расположен в определенном участке хромосомы, и в диплоидной фазе жизненного цикла человека и других метазоа есть две хромосомы с этим участком гена в каждом ядре. Эти две копии гена называются аллелями. Особенно актуально для этого отчета то, что многие вариантные аллели каждого гена возникли в ходе эволюции человека, и разные аллели часто дают небольшие или большие различия в некоторых конкретных признаках организма при сравнении членов популяции.
В этой главе комитет описывает области генетики и геномики человека.Обсуждается роль молекулярной эпидемиологии в обнаружении токсических веществ, вызывающих развитие, а также трудности в обнаружении сложных взаимодействий генотип-среда.
ГЕНОТИП, ФЕНОТИП И МНОГОФАКТОРИАЛЬНОЕ НАСЛЕДОВАНИЕ
В своих классических экспериментах середины девятнадцатого века Грегор Мендель (1865) выбрал растение гороха ( Pisum sativum ) для изучения сегрегации и набора специфических детерминант фенотипа. черты. Ему посчастливилось выбрать несколько признаков, каждая из которых контролировалась одним генетическим локусом.Аллели в каждом локусе при наследовании действовали либо доминантным, либо рецессивным образом, и в условиях его тестирования на их действие не оказывали значительного влияния другие гены или факторы окружающей среды. Следовательно, он наблюдал точные и интерпретируемые математические соотношения фенотипов потомства в каждом эксперименте по разведению. Признаки фенотипа, которые демонстрируют такие легко интерпретируемые образцы наследования, называются простыми, или менделевскими, признаками, и они обычно регулируются одним генетическим локусом.
Однако связь между генотипом и фенотипом почти всегда очень сложна. Даже когда ученые рассматривают один конкретный ген и знают его конкретную аллельную форму, его влияние на фенотип часто зависит от одной или обеих из двух переменных: (1) различных аллелей различных других основных генов и генов-модификаторов в геноме организма и (2) ) различные условия окружающей среды. Такие признаки демонстрируют многофакторный паттерн наследования (также называемый сложным или неменделевским наследованием) и называются сложными признаками или мультиплексными фенотипами (недавний обзор см. Lander and Schork 1994).Многофакторное наследование встречается гораздо чаще, чем простое наследование. Такие признаки влекут за собой взаимодействие двух или более генов (полигенный признак). Гены могут вносить вклад в фенотипический признак количественным и аддитивным образом (например, гены A , B и C могут вносить 20%, 30% и 50%, соответственно, в такой признак, как рождение ребенка). масса). Эти гены называются «локусами количественных признаков», и генетические методы анализа их вклада очень эффективны.Например, аллели гена BRCA1 , по-видимому, вносят около 5% в общий риск рака груди (для обзора см. Brody and Biesecker 1998), но несколько других участников, которые, как считается аналитически, существуют, не пока не выявлено. Еще более сложные закономерности наследования можно проследить до нескольких генов, действующих неаддитивным образом. Сегрегированные аллели могут быть ни доминантными, ни рецессивными. Наконец, ген может проявлять неполную пенетрантность (только некоторые члены популяции проявляют признак) или переменную экспрессивность (члены популяции различаются по степени признака) или и то, и другое.
Другие черты могут быть изменены окружением. Такие признаки вовсе не необычны и могут пересекаться с полигенными признаками. Исследования на модельных организмах, таких как плодовая муха Drosophila melanogaster , давно показали, что влияние гена на признак может быть изменено такими внешними факторами, как температура, химические вещества, питание и теснота. Генетики, особенно интересующиеся эволюцией, утверждали, что взаимодействия генов с окружающей средой настолько распространены и важны, что следует говорить не о «фенотипическом признаке» организма, а о его «норме реакции», которая представляет собой набор фенотипов, производимых индивидуальный генотип, когда он подвергается воздействию различных условий окружающей среды (Stearnes, 1989).Связь между генотипом, средой и фенотипом, которую иногда называют взаимодействием ген-среда, можно выразить как
Генотип + Среда → Фенотип.
Хотя многофакторная наследственность доставляет неудобства генетикам, она описывает большинство наследственных заболеваний человека и практически все уязвимости. Как упоминалось в главе 2, примерно 25% дефектов развития человека, возможно, являются следствием многофакторного наследования. Общеизвестно, что люди и экспериментальные животные неоднородны в своей реакции на лекарства или загрязнители окружающей среды.В настоящее время предпочтительное объяснение состоит в том, что неоднородность отражает комбинацию гетерогенных обстоятельств воздействия (внешние условия) и гетерогенных генотипов восприимчивости (внутренние условия). Примерами воздействия плюс восприимчивости могут быть возраст начала рака легких у курильщиков сигарет или вероятность астмы, вызванной загрязнением в городах. Взаимосвязь между геном и окружающей средой в токсикологии развития еще более усложняется необходимостью учитывать генотип как матери, так и эмбриона или плода, как и где метаболизируется токсикант, а также стадию развития, на которой токсикант проникает через плаценту.Взаимодействие генов с окружающей средой, очевидно, имеет отношение к областям молекулярной эпидемиологии и токсикологии развития.
ПОЛИМОРФИЗМЫ
Полиморфизм означает наличие двух или более аллелей определенного гена в популяции организмов; аллель меньшинства присутствует с частотой гена не менее 1% (Hartl and Taubes, 1998). Эта частота является несколько произвольным порогом, установленным популяционными генетиками, а аллели меньшинств с еще более низкой частотой называются «редкими аллелями».В соответствии с распределением Харди-Вайнберга (p 2 + 2pq + q 2 ) для двух аллелей в одном локусе, если меньшинство аллелей присутствует с частотой гена 1%, тогда он присутствует в гетерозиготной форме. примерно у 2% представителей этой популяции и в гомозиготной форме у 0,01% населения.
Какой бы ни была частота, аллели теперь определяются в самом общем виде, а именно как разные нуклеотидные последовательности одного и того же гена, то есть как изменения одного или нескольких оснований (аденина, тимина, цитозина и гуанина) относительно эталонная последовательность оснований ДНК.Однако обнаружение такой разницы само по себе мало что говорит о влиянии на фенотип. Если разница в последовательности возникает в кодирующей области гена, это может повлиять на активность или стабильность белка. Если изменение является синонимичным (т. Е. Аминокислота не изменена), консервативным или локализованным в области белка, где допустима любая из нескольких аминокислот, активность или стабильность белка могут не пострадать. Если изменение последовательности ДНК происходит в транскрибируемой области гена, но не в кодирующей области, это может повлиять на рамку считывания, сплайсинг, стабильность мРНК, эффективность трансляции или регуляцию транскрипции.Если находится вне области транскрипции гена, изменение все же может повлиять на время, место и уровень экспрессии гена, но не на последовательность белка. Необходимо провести дополнительную работу, чтобы определить влияние конкретного изменения ДНК на функцию или уровень белка.
Обычно ожидается, что полиморфизмы, достигающие уровня аллельной частоты 1%, будут иметь селективно благоприятные фенотипические последствия изменения активности или количества белка. Однако вариант может быть представлен ниже уровня 1% или близкого к 1%, потому что он возник в небольшой группе организмов-основателей, которые из-за местных случайных обстоятельств распространились на большую популяцию по сравнению с другими представителями этого вида.Такой полиморфизм может не влиять на активность или количество белка. Это было бы просто маркером этой линии организмов. Это может даже иметь отрицательный селективный эффект.
Современные методы секвенирования значительно расширили возможности исследователей по обнаружению аллелей. Для конкретной генной последовательности любые два неродственных человека в популяции, вероятно, будут иметь различия в последовательностях. Предполагается, что последовательность гена включает все регуляторные и транскрибируемые области ДНК. Когда сначала возникает изменение основания из-за окислительных ударов, ошибок репликации, индуцированных ультрафиолетом димеров тимина или других форм повреждения ДНК, обычно требуется один цикл синтеза новой ДНК, чтобы стать «закрепленным» в двухцепочечной форме, которая невосприимчива к ремонт и считаться мутацией.Перед этим синтезом изменение основания часто приводит к несоответствию двойной спирали ДНК, и ряд ферментов репарации несоответствия устраняют такие ошибки (Snow 1997). Однако из каждого миллиона или более участков ДНК, которые были повреждены, ошибка иногда остается неисправленной. Считается, что невосстановленные мутации возникают естественным образом с частотой один раз на 10 6 -10 8 оснований на поколение. Поскольку у людей такой большой геном, у каждого человека за всю жизнь накапливается примерно 75 новых мутаций.Большинство из них, вероятно, не вредны. Многие из них не встречаются в областях, кодирующих белок (5% последовательности ДНК человека), или, если они встречаются, не изменяют конкретную аминокислоту (синонимичные замены). Некоторые из них, однако, вредны, а частота вредных мутаций у людей (несинонимичные изменения аминокислот, влияющие на активность), по недавним оценкам, составляет не менее 1,6 новых вредных мутаций на диплоидный геном на поколение. Авторы приходят к выводу, что этот показатель «близок к максимальному допустимому уровню для таких видов, как люди, у которых низкий уровень воспроизводства» (Eyre-Walker and Keightley 1999).Вероятно, что человеческая популяция полна генетических вариаций, и эти вариации должны рассматриваться и оцениваться при любой оценке индивидуальной восприимчивости к токсическим веществам, связанным с развитием.
ПРОЕКТ ЧЕЛОВЕЧЕСКОГО ГЕНОМА
Геном организма — это общее генетическое содержимое организма, или, в более широком смысле, это полное содержание ДНК организма, включая нетранскрибируемые, не- цис- -регулирующие области ДНК, такие как центромеры и теломеры. Изучение геномов организмов, которое называется геномикой, включает области исследований, определяющие генетические и физические карты геномов, последовательности ДНК геномов, функции генов и белков, регулирующие элементы генов cis и время, место и условия экспрессии генов.Важной частью геномики стало управление огромным объемом собранной информации (область, называемая биоинформатикой) и анализ данных, касающихся, например, аспектов организации генома, сравнения геномов различных организмов и глобальные паттерны экспрессии генов.
Проект генома человека (HGP) был запущен в октябре 1990 года Национальными институтами здравоохранения (NIH) в качестве инициативы, финансируемой из федерального бюджета. Непосредственной целью тогда, как и сейчас, было завершение точного определения последовательности приблизительно 3.5 миллиардов пар оснований ДНК человека (количество гаплоидов) к концу 2003 г. (F.S. Collins et al. 1998). «Предварительный набросок последовательности», составляющий примерно 90% генома человека, был завершен в середине 2000 г. (www.ornl.gov/hgmis/project/progress.html). В более долгосрочной перспективе цель состоит в том, чтобы идентифицировать все гены человека. Идентификация затруднена. В таком организме, как дрожжи, который благоприятен для идентификации генов с помощью мутационного генетического анализа, более половины генов оставались необнаруженными до тех пор, пока не стала доступна последовательность генома (Brown and Botstein 1999).Отсутствие обнаружения было частично из-за больших повторяющихся областей генома дрожжей. У позвоночных мутационный генетический анализ намного сложнее, и дублирование может быть более распространенным. Таким образом, предпочтительным подходом является первоначальная идентификация гена путем секвенирования. Ген первоначально идентифицируется как открытая рамка считывания (ORF), которую можно определить непосредственно, глядя на последовательность, или он первоначально идентифицируется как сайт экспрессируемой метки последовательности (EST), последовательность, комплементарная известному фрагменту транскрибируемой РНК. (увидеть ниже).После этого цель состоит в том, чтобы идентифицировать каждый ген как последовательность, кодирующую полноразмерную РНК и белок с известной функцией. Также необходимо будет выяснить функции нетранскрибируемых областей, таких как многочисленные большие цис -регуляторные области, устанавливающие условия для экспрессии генов. Эта задача еще более сложна и в настоящее время включает ряд подходов, включая создание линий трансгенных животных, несущих части регуляторной области в сочетании с репортерным геном (например,g., зеленый флуоресцентный белок, GFP).
Функциональный анализ генома с точки зрения времени и места экспрессии генов и функций генных продуктов иногда называют «функциональной геномикой» или даже «постгеномикой». Анализ функции белка может быть простым, если последовательность белка похожа на последовательность другого хорошо изученного белка, и может быть трудным, если мотивы последовательности отсутствуют. Анализ белкового состава организма (называемого «протеом») и функции иногда называют «протеомикой».«Целевые области HGP в настоящее время включают генетическое и физическое картирование генома человека, секвенирование ДНК, анализ геномов многих важных нечеловеческих организмов, информатику для обработки огромного увеличения скорости генерирования информации, развитие ресурсов и технологий, а также этические, правовые и социальные последствия (ELSI) генетических исследований для людей и общества (FS Collins et al. 1998).
HGP поддерживается NIH и Министерством энергетики США (DOE) в 22 специализированных исследовательских центрах генома в США и во многих университетских, национальных и частных лабораториях.В NIH название было изменено на Национальный центр исследований генома человека в 1993 году, а с конца 1996 года он стал Национальным исследовательским институтом генома человека (NHGRI). По крайней мере, 14 других стран также имеют программы по анализу геномов различных организмов — от микробов и экономически важных растений и животных до людей.
Взрыв информации о геномике произошел раньше, чем предсказывали самые смелые ученые. После первой полной последовательности генома, Haemophilus influenzae в 1995 году, в следующие 18 месяцев были завершены еще семь геномов, а именно, еще четыре генома эубактерий, два генома архебактерий и один геном одноклеточных эукариот — генома дрожжей Saccharomyces cerevisiae .В декабре 1998 г. был завершен геном первого многоклеточного эукариота, Caenorhabditis elegans (100 килобайт последовательности ДНК и 19000 генов, идентифицированных, по крайней мере, как открытые рамки считывания). По состоянию на конец 1999 г. было частично идентифицировано, локализовано и секвенировано более 30 000 генов человека. 22-я хромосома человека была секвенирована и, по прогнозам, содержит по крайней мере 679 генов (Dunham et al. 1999). У мышей описано не менее 14000 генов. Последовательность дрозофилы ( Drosophila melanogaster ) была завершена в 1999 г. (Adams et al.2000). В середине 2000 года была завершена приблизительная («рабочая черновая») последовательность действий человека. К 2003 году также будут секвенированы многочисленные нечеловеческие геномы, в том числе мышь Mus musculus , рыба данио Danio rerio , шелкопряд Bombyx mori , крыса, собака, кошка, курица, рис, кукуруза, пшеница, ячмень. , хлопок, растение Arabidopsis thaliana , а также, вероятно, корова, овца, свинья и лошадь. Секвенирование генома мыши идет с опережением графика.
Новые технологии, ресурсы и приложения становятся все более доступными для исследователей из многих различных научных областей, включая исследования рака, открытие лекарств, медицинскую генетику и генетику окружающей среды, и их доступность также должна ускорить многочисленные важные достижения в токсикологии развития в следующем десятилетие, как обсуждается далее в этой главе.
Функциональная геномика и технология микрочипов
С самого начала ожидалось, что завершение секвенирования генома человека станет лишь первым шагом в HGP.Информация о последовательности и расположении генов в геноме значительно облегчит дальнейшие исследования — не только генетической изменчивости человека, но и функциональной геномики. Как отмечалось выше, последний представляет собой всесторонний анализ экспрессии гена и функции генного продукта. В случае дрожжей и C. elegans , для которых уже известна вся последовательность генома, в настоящее время ведутся проекты по систематической оценке функции каждого генного продукта, например, путем выключения генов дрожжей (вызывая потерю функции). по одному и путем связывания идентифицированной матричной (м) РНК с каждой открытой рамкой считывания.
Некоторые из этих функциональных анализов можно продолжить даже до секвенирования генома. В случае людей изучение EST было важным этапом такого анализа. мРНК могут быть выделены из организма и преобразованы в комплементарные (в) ДНК с помощью обратной транскрипции (ОТ) и полимеразной цепной реакции (ПЦР). Затем кДНК клонируют и секвенируют для получения большой и четко определенной библиотеки EST. Информация заносится в базу данных. Эти последовательности представляют собой гены, экспрессируемые у человека.Например, сейчас доступно более 1 миллиона человеческих EST, представляющих более 50 000 генов. Каждый EST отражает часть мРНК, а не полноразмерную последовательность. Наиболее полные библиотеки готовятся из широкого диапазона тканей и времен развития, чтобы включить все экспрессируемые гены. (К сожалению для токсикологов развития, хотя исходные источники РНК включали плаценту, они были недостаточно представлены в разнообразии ранней эмбриональной ткани.) Эти последовательности полезны в ходе секвенирования генома для идентификации участков ДНК, которые фактически кодируют белки (только 5% от считается, что последовательность генома человека проявляется в процессированных последовательностях мРНК).Появились новые методы получения полноразмерных кДНК из транскриптов, и они будут более полезными, чем фрагменты.
Следующим этапом анализа функции генома является определение времени, места и условий экспрессии каждого гена. До недавнего времени этот анализ проводился по одному гену. Методы микроматрицы ДНК недавно сделали возможным описание одновременных изменений тысяч генов по мере развития клеток и тканей или различных изменений условий окружающей среды.При изучении токсического воздействия на организм анализ иногда называют «токсикогеномикой» или, при изучении воздействия фармацевтических препаратов, «фармакогеномикой». Подходы с ДНК-микрочипами получают широкое распространение (обсуждение его использования в токсикологии см. В Nuwaysir et al. 1999).
Теперь технология пригодна для одновременного сравнения количества тысяч видов мРНК в двух тканях или образцах клеток (например, нормальная контрольная ткань и ткань, обработанная тератогенным агентом).Для сравнения тысячи различных последовательностей ДНК (например, каждая олигомер из не менее 25 нуклеотидов) автоматически наносятся на микропрепарат, и каждая последовательность помещается в известное место для создания микроматрицы ДНК. ДНК прилипает к стеклу, и каждое пятно ДНК обычно имеет диаметр 20 микрометров (м). Например, микроматрицы из 6200 последовательностей кДНК, представляющих все гены дрожжей, были помещены на одном слайде или нескольких слайдах, а 8900 последовательностей кДНК, представляющих около 10% генов человека (в основном последовательности EST), были помещены на несколько слайдов. слайды (Iyer et al.1999). Стоимость и время изготовления таких слайдов достаточно скромны, чтобы можно было приготовить сотню или около того, каждый из которых служит для анализа одного условия сравнения (например, анализа нескольких временных точек и условий концентрации). Каждый слайд представляет собой набор «зондов» ДНК, с помощью которых количество тысяч видов мРНК в клетках, тканях или эмбрионах может быть визуализировано одновременно в каждый момент времени и при любом условии.
В соответствии с процедурой Brown и Botstein (1999) образцы тканей для сравнения извлекаются отдельно, и мРНК помечаются различными молекулами флуоресцентного красителя, например, зеленым для контроля и красным для обработанной ткани (см.).Затем РНК смешивают и добавляют на предметное стекло микроматрицы (в их случае, несущее 8900 последовательностей ДНК человека) в условиях, подходящих для гибридизации РНК-ДНК, а затем промывают для удаления несвязанной РНК. Затем слайд просматривают в флуоресцентном микроскопе, чтобы увидеть, связывает ли каждое конкретное пятно ДНК больше зеленого или рРНК. Отношение красного к зеленому говорит о том, экспрессируется ли конкретный ген в большей или меньшей степени, чем обычно, в условиях лечения. Желтый цвет отображается, когда гибридизуются одинаковые красные и зеленые мРНК.Этот метод был недавно применен к человеческим клеткам, культивируемым в присутствии или в отсутствие сыворотки. Действительно, сотни генов изменили экспрессию, в том числе многие гены, кодирующие связанные со стрессом белки, наблюдаемые при заживлении ран (Iyer et al. 1998), факт, который ранее не осознавался в годы изучения сывороточного ответа культивируемых клеток. Этот метод также применялся к дрожжевым клеткам, прогрессирующим по пути споруляции (Chu et al. 1998), дрожжевым клеткам, проходящим через клеточный цикл (Spellman et al.1998) и дрожжевые клетки в гаплоидном, диплоидном или тетраплоидном состоянии (Galitski et al. 1999). Недавно его использовали для обнаружения ответа одного вида клеток на два сигнальных лиганда, каждый из которых действует через разные рецепторные тирозинкиназы (Fambrough et al. 1999). Во всех случаях изменялась экспрессия сотен генов. Просмотр глобальных закономерностей показывает, что каждый ген не ведет себя индивидуально; вместо этого согласованная экспрессия больших батарей генов, по-видимому, происходит в различных условиях.Эти результаты подтверждают ценность анализа глобальных паттернов экспрессии генов. Предыдущие исследования отдельных генов могли упустить крупномасштабные закономерности. Однако многое еще предстоит сделать для интерпретации разнообразных изменений экспрессии генов. Как упоминалось выше, наблюдаются сотни изменений экспрессии генов даже при, казалось бы, скромных изменениях в условиях клетки, таких как ее плоидность.
РИСУНОК 5-1
ДНК-микрочипы как средство для одновременного определения количества тысяч видов мРНК в клетке или ткани.Как показано в верхнем левом углу, мРНК получают отдельно из двух типов клеток, таких как нормальные и опухолевые, и каждая мРНК преобразуется (подробнее …)
При анализе токсических эффектов ожидается, что клетки или организмы можно было лечить токсичными веществами с неизвестным механизмом действия, а изменения экспрессии генов можно было профилировать с помощью метода ДНК-микрочипов. Если бы было достаточно известно о функции и взаимодействии белков, кодируемых генами, претерпевающими изменения экспрессии, можно было бы сделать обоснованные выводы о механизме действия токсиканта.Ожидается, что в будущем методы микроматрицы ДНК позволят быстро и подробно охарактеризовать реакцию клетки или организма на токсикант. По мере сбора большего количества информации различные токсиканты могут быть сгруппированы по сходству их действия, и анализ токсического действия может проводиться на более систематической основе. Метод микроматрицы ДНК уже используется для сравнения нормальных и раковых клеток. В этом случае также исследуются многие вопросы интерпретации различий в экспрессии генов.
Хотя предпочтительно иметь большой набор клонированных и секвенированных ДНК (представляющих различные идентифицированные гены) для микроматрицы, как в случае дрожжей, в этом нет необходимости. EST уже были полезны для исследований на людях и мышах, как в упомянутом выше исследовании сыворотки (Iyer et al. 1999). Если обнаруживается, что экспрессия конкретной последовательности, известной только по ее EST, сильно изменяется в условиях тестирования, она может быть квалифицирована как достаточно интересная, чтобы заслужить клонирование полной длины, секвенирование и дальнейший анализ функции.
При таких сравнениях накапливается огромное количество данных (например, когда 6200 дрожжевых генов (весь дрожжевой геном) или 8900 человеческих последовательностей экспрессируются по-разному в нескольких условиях в разные моменты времени). Многомерные наборы данных поставили перед прикладными математиками задачу найти средства для их выражения способами, полезными для биологов (например, см. Методы двумерного кластерного анализа в Eisen et al. 1998; Alon et al. 1999; Tamayo et al. 1999). . Тем не менее, на горизонте маячат большие наборы данных (например,g., экспрессия, возможно, 100 000 генов мыши или человека во все времена и в любом месте развития, не говоря уже о воздействии различных токсичных веществ). Как описано ниже, менеджеры и аналитики пользуются большим спросом на эти наборы данных.
Хотя различные методы микроматрицы обещают раскрыть захватывающую новую информацию о том, где, когда и при каких условиях транскрибируются гены генома, этот подход не предоставит информации о трансляции и посттрансляционной модификации белков, кодируемых этими мРНК. — то есть информация о том, когда и где белки присутствуют и активны.Функция белков почти всегда является непосредственной причиной функционирования клеток. Предоставление такой функциональной информации — цель протеомики. Протеомика была определена (Андерсон и Андерсон, 1998) как «использование количественных измерений экспрессии генов на уровне белка для характеристики биологических процессов (например, болезненных процессов и эффектов лекарств) и расшифровки механизмов контроля экспрессии генов». В основе протеомики лежит индекс человеческого белка, то есть систематическая идентификация всех белков человека (Anderson and Anderson, 1982) с использованием высокопроизводительного, двухмерного (2D) -гелевого электрофореза с высоким разрешением для создания геля с на нем целых 1000 отдельных белковых пятен.Большой объем информации о 2D-геле хранится в базе данных Proteomics (адрес в Интернете см. В Приложении B). Были разработаны планы идентифицировать каждое белковое пятно на 2D-геле, потому что количество белка в нанограммах в пятне достаточно для определения частичной аминокислотной последовательности с помощью тандемной масс-спектрометрии (Yates 1998). Затем частичную последовательность можно найти в базе данных генома и идентифицировать белок. Когда белки модифицируются фосфорилированием, ацилированием, гликозилированием, фарнезилированием, ограниченным протеолизом или любым из других 30 или около того ковалентных посттрансляционных изменений, их миграция на 2D-геле изменяется, что позволяет установить корреляцию с их активностью или неактивностью. в ткани.Такую информацию об активности нельзя получить из измерений количества мРНК с помощью микроматрицы ДНК.
Наконец, многие белки должны связываться с другими белками для достижения активности, и существуют различные неденатурирующие методы гель-электрофореза для обнаружения таких ассоциаций. Можно ожидать, что текущие и будущие усилия приведут к новым технологическим достижениям в области анализа белков и их функций. В токсикологии развития сочетание геномики и протеомики дает возможность оценивать токсиканты развития не только по их способности изменять экспрессию генов, но также по их способности изменять функцию белков.
Применение геномных технологий
Недавно исследователи использовали геномные технологии для идентификации и определения последовательности генов, играющих роль в этиологии заболевания. Вероятно, что эти геномные технологии будут применены для изучения воздействия химических веществ на развитие в ближайшем будущем. Уже существуют две модели, демонстрирующие применение новых геномных технологий: проект анатомии генома рака (CGAP), который начался в 1997 году и осуществляется Национальным институтом рака, и проект экологического генома (EGP), который начался в 1998 году и является под управлением Национального института наук об окружающей среде.
Цель CGAP — предоставить полный каталог всех генов, экспрессия которых изменяется в раковых клетках относительно нормальных клеток для всех типов рака (Pennisi 1997). В прошлом основным препятствием для анализа раковых клеток была смесь типов клеток (нормальных и злокачественных), присутствующих в типичной опухоли. Трудно получить точную картину экспрессии генов в раковых клетках, если РНК, выделенная из всей опухоли, включает последовательности из многих различных типов клеток.Используя недавно разработанные методы, исследователи CGAP минимизируют эту проблему, выбирая под микроскопом небольшую однородную группу клеток, нормальных, предраковых или злокачественных (Emmert-Buck et al. 1996; Simone et al. 1998), которые затем изолируются путем прикрепления к ним. на специальную лазерно-чувствительную пленку. Затем последовательности РНК извлекаются из изолированных клеток и амплифицируются с помощью ОТ-ПЦР для создания библиотек кДНК (Peterson et al. 1998). Для получения широкого представления мРНК потребовалось около 5000 клеток гомогенной морфологии.Для получения кДНК многочисленных видов мРНК необходимо всего 500 клеток. Такие библиотеки будут важными инструментами для описания прогрессирования рака и предоставления диагностических маркеров заболевания. Они также предоставляют последовательности для функционального анализа с помощью микрочипов ДНК. По мере получения данных они вводятся на веб-сайт CGAP для быстрого доступа и использования другими исследователями, а также для интеграции с другими данными в большом репозитории.
Ожидается, что новые технологии сократят минимальное количество необходимых клеток до 1000 или меньше и по-прежнему получат широкое представление РНК.Чем ниже минимальное необходимое количество клеток, тем лучше для будущих токсикологических исследований развития, поскольку многие развивающиеся типы клеток присутствуют в небольших количествах во время эмбриогенеза и внутриутробного развития плода.
Целью EGP является изучение взаимодействия между генетической восприимчивостью, воздействием окружающей среды и болезнями. В частности, исследователи, работающие над этим 5-летним проектом, пытаются идентифицировать аллельные варианты (т.е. полиморфизмы) 200 генов, важных при экологических заболеваниях, разработать централизованную базу данных полиморфизмов для этих генов и способствовать эпидемиологическим исследованиям взаимодействия генов с окружающей средой. в этиологии заболевания.Как обсуждается далее в этой главе, доказательства особенно убедительны в пользу повышенной химической восприимчивости людей с полиморфизмом генов, кодирующих ферменты, метаболизирующие лекарственные средства (DME). EGP использует геномные технологии, такие как высокопроизводительный анализ последовательностей, разработанный для HGP, который облегчит эпидемиологические исследования взаимодействия генов и окружающей среды при заболеваниях.
Ожидается, что тип геномных технологий, используемых в CGAP и EGP, и многие из самих данных окажут огромное влияние на будущие исследования в области токсикологии развития.В идеале аналог токсикологии развития таких программ, как CGAP и EGP, должен был бы сосредоточиться на получении данных об экспрессии генов в любое время и во всех тканях во время нормального развития. Затем можно было провести сравнение эмбрионов и плодов беременных контрольных (нормальных) животных и беременных животных, получавших лечение, для поиска различий в экспрессии генов. Изменения могут быть идентифицированы в экспрессии генов, кодирующих белки, которые, как известно, функционируют в клеточной передаче сигналов, регуляции транскрипции, делении клеток, подвижности клеток, клеточной адгезии, апоптозе, дифференцировке, метаболизме, восстановлении, электролитном балансе, гомеостазе или транспорте.Такие исследования помогут выяснить механизмы, с помощью которых внешние химические вещества (потенциальные токсиканты развития) действуют как агонисты или антагонисты рецепторных и ферментно-опосредованных субклеточных процессов во время эмбриогенеза и внутриутробного развития плода. Такие исследования станут важной силой в объединении областей биологии развития, геномики и токсикологии развития.
Управление последовательностью генома и данными функциональной геномики
Взрыв молекулярной биологии за последние два десятилетия привел к огромным успехам в секвенировании ДНК, что, в свою очередь, привело к все более быстрой идентификации генов как ORF и как сайты EST и идентификация функции генных продуктов по мотивам последовательности (например,g., гомеодомены, домены цинковых пальцев, домены киназы и домены Sh3 и Sh4). Существует четыре основных базы данных нуклеотидных последовательностей: GenBank и База данных геномных последовательностей (GSDB) в США, Европейская библиотека молекулярной биологии (EMBL) и Банк данных ДНК Японии (DDBJ). Все группы обмениваются новыми и обновленными последовательностями в электронном виде и обычно в один и тот же день подачи.
История этих баз данных насчитывает два десятилетия. Целью Лос-Аламосской библиотеки последовательностей в 1979 году в Лос-Аламосской национальной лаборатории (LANL), спонсируемой Министерством энергетики (DOE), было хранение данных последовательностей ДНК в электронной форме.В том же году аналогичная база данных была создана в EMBL в Гейдельберге. В 1982 году было решено, что любые данные, представленные или введенные одной группой, будут немедленно пересылаться другой, что позволит избежать дублирования усилий. В 1982 году база данных LANL стала GenBank, когда Bolt, Beranek and Newman (BBN) стал основным подрядчиком по распределению поддержки данных и пользователей, а LANL был изменен на субподрядчика BBN. Деятельность BBN и LANL по работе с данными последовательностей спонсировалась Национальным институтом общих медицинских наук (NIGMS), а также Министерством энергетики и другими агентствами.По окончании первого 5-летнего контракта IntelliGenetics стала основным подрядчиком, а LANL снова стала субподрядчиком, отвечающим за проектирование и создание базы данных. В октябре 1992 года, по окончании второго пятилетнего контракта, NIGMS передала GenBank Национальному центру биотехнологической информации (NCBI) при Национальной медицинской библиотеке. В августе 1993 года ресурсы баз данных LANL и NCBI стали независимыми друг от друга. GenBank остался в NCBI, а LANL взяла новое название База данных геномных последовательностей (GSDB) и переехала в Национальный центр геномных ресурсов (NCGR) в Санта-Фе, Нью-Мексико.База данных нуклеотидных последовательностей EMBL, созданная в 1982 году, в настоящее время поддерживается Европейским институтом биоинформатики (EBI), расположенным недалеко от Кембриджа, Англия, который также курирует базу данных последовательностей белков SWISS-PROT и более 30 других специализированных баз данных. DDBJ, созданный в 1984 году и спонсируемый Министерством образования, науки и культуры Японии с 1986 года, накапливает данные о нуклеотидных последовательностях, в основном от японских ученых, и посредством электронной передачи делает доступными более десятка других баз данных.
В течение десятилетия будут секвенированы геномы по крайней мере 200 организмов, от многочисленных видов бактерий до людей. К тому времени анализы экспрессии с использованием высокопроизводительных микрочипов ДНК, кДНК или белковых микромассивов станут обычным явлением и предоставят нам огромное количество новой информации о времени и месте (то есть тканях и типах клеток) экспрессии различных генов и о изменения экспрессии генов в развитии организма и реакция на различные условия воздействия (Reichhardt 1999).Будет огромная потребность в отделах или отделах биоинформатики в университетах и отраслях, чтобы отслеживать данные и анализировать их в отношении интересных вопросов об организации и функции генома (см. Комментарий Reichhardt 1999). Дополнительная информация, вероятно, появится при сравнении геномов разных организмов. Потребность в обучении большого количества людей новой области биоинформатики будет огромной. Информация, легко доступная в Интернете, должна способствовать интеграции областей токсикологии развития, генетики человека, геномики и биологии развития.В будущем токсикологи, занимающиеся вопросами развития, безусловно, получат множество преимуществ от быстрого и немедленного доступа к этой новой информации, но следует понимать, что потребуется обучение, прежде чем можно будет эффективно использовать огромные объемы информации. В настоящее время также не определено, как лучше всего организовать данные, чтобы лица, участвующие в оценке риска, могли получить наиболее актуальные.
ПОСЛЕДНИЕ СОБЫТИЯ МОЛЕКУЛЯРНОЙ ЭПИДЕМИОЛОГИИ
В последнее время молекулярные инструменты были использованы для выявления взаимодействий между генетическими факторами и факторами окружающей среды, вызывающими сложные заболевания, такие как дефекты развития.Большинство неблагоприятных исходов беременности у людей имеют неизвестную этиологию и рассматриваются как сложные черты, при которых экзогенные агенты могут взаимодействовать с определенными комбинациями аллельных вариантов генов, контролирующих развитие и дифференцировку, что приводит к неблагоприятным исходам беременности. Исследования взаимодействий генов и окружающей среды во время эмбриогенеза и развития плода стали обычным явлением и получили все большее признание. Категории этиологии болезни можно рассматривать как охватывающие диапазон от полностью генетической причины до полностью окружающей среды.Эти категории включают причинность одного гена, хромосомную причинность, многофакторную причинность с высокой наследуемостью, многофакторную причинность с низкой наследуемостью, инфекционные причины и причины окружающей среды (Khoury et al. 1993a).
Многие расстройства с одним геном характеризуются низкой частотой аллеля болезни в общей популяции (частота аллелей 1% или меньше) и высокой пенетрантностью (заболевание развивается у большой доли людей с аллелем болезни). Гены предрасположенности к этим моногенным нарушениям обычно демонстрируют менделевские закономерности наследования и связаны с высоким риском заболевания.Хотя по отдельности они редки, менделевские расстройства с одним геном вносят значительный вклад в младенческую заболеваемость и смертность. Примерно 4-7% госпитализаций в педиатрические больницы приходится на признанные менделевские заболевания (Khoury et al. 1993b). Из более чем 2000 вероятных синдромов дефекта развития с одним геном у людей ген был выделен и картирован для 100 из этих синдромов и картирован, но еще не выделен для еще 100 (Winter 1996). Для сравнения, у мышей имеется примерно 500 спонтанно возникающих дефектов одного гена, связанных с дефектами развития.Приблизительно 75 из этих генов были выделены и более 400 картированы (Winter 1996). Более того, было приготовлено более 1000 мутантов мышей с известными дефектами генов (многие из которых относятся к компонентам сигнальных путей), и многие из них имеют фенотипы, которые полностью квалифицируют их как дефекты развития одного гена мыши. Несмотря на наличие мутантов мышей и идентификацию генов, важных для развития C. elegans , Drosophila и рыбок данио (см. Главы 6 и 7), изучение дефектов одного гена, способствующих дефектам развития у людей, не привело к тем не менее получил много экспериментального внимания.Гены, идентифицированные как важные для развития у C. elegans , Drosophila и рыбок данио, однако, предоставляют богатый источник информации для идентификации генов потенциальной восприимчивости у людей. Это поражает этот комитет как недоиспользуемый ресурс.
Многофакторные расстройства или сложные заболевания характеризуются генетической сложностью и вероятными взаимодействиями между генами и окружающей средой (Ellsworth et al. 1997). Эти болезни, как правило, накапливаются в семьях, но не наследуются простым менделевским способом.Обычно они встречаются в большей степени в затронутых семьях, чем ожидалось в общей популяции. В отличие от заболеваний, связанных с одним геном, гены предрасположенности к сложным расстройствам обычно встречаются в популяции (частота аллелей более 1%) и могут считаться полиморфизмами. Гены восприимчивости обычно связаны с низким риском для индивидуума, но высоким риском в популяции. В случаях высокой наследуемости признак заболевания может иметь аллели нескольких основных генов и нескольких генов-модификаторов, способствующих его пенетрантности и экспрессивности.У людей с низкой наследуемостью могут быть аллели нескольких генов, а также определенные факторы окружающей среды, которые вместе увеличивают риск заболевания в популяции.
Недавние примеры, в которых взаимодействия генов с окружающей средой были успешно выяснены для дефектов развития, следующие:
Полиморфизм трансформирующего фактора роста ( TGF α) и расщелины ротовой полости: Доказательства связи между курением матери и расщелинами полости рта имеют были двусмысленными (Hwang et al.1995). В этом исследовании не было общей значимой связи между курением матери и расщелинами ротовой полости у новорожденных. Однако, если у новорожденного был вариантный аллель ( TAQL C2) гена TGF α, отношение шансов для расщелины ротовой полости у грудных детей от курящих матерей (более 10 сигарет в день) составляло 8,7, что в 10 раз больше. по сравнению с младенцами от курящих матерей, у которых не было этого вариантного аллеля. Вариантный аллель сам по себе не был связан с повышенным риском ротовой щели. TGF α является лигандом рецептора тирозинкиназы.
- Ген гомеобокса MSX1 , дефекты конечностей и курение: Частоты редких аллелей в локусе MSX1 немного выше у младенцев с дефектами конечностей по сравнению с младенцами с другими типами пороков развития (отношение шансов 2,4). Младенцы, несущие редкие аллели, имели в 2 раза повышенный риск дефицита конечностей, когда мать курила во время беременности (отношение шансов 4,8), по сравнению с младенцами, несущими редкий аллель, матери которых не курили.Само по себе курение не было связано с повышенным риском дефектов конечностей в этом исследовании (Hwang et al. 1998). MSX1 — это фактор транскрипции, активность которого часто зависит от сигналов BMP2,4.
- Различная предрасположенность человека к порокам развития из-за дифенилгидантоина (DPH или фенитоина): 10-20% потомков женщин с эпилепсией, принимающих фенитоин во время беременности, имеют фетальный гидантоиновый синдром (Hanson et al. 1976; van Dyke et al. 1988) . Считается, что фенитоин превращается в реактивный промежуточный продукт, оказывающий тератогенное действие (Martz et al.1977; для обзора см. Finnell et al. 1997а). Изменчивость популяции в ответ на фенитоин, возможно, отражает гетерогенность генотипов DME. В паре близнецов, в которых только один близнец имел дисморфологию гидантоинового синдрома, у матери и пострадавшего близнеца была снижена активность фермента эпоксидгидроксилазы по сравнению с здоровым близнецом (Buehler 1984). Бюлер и др. (1990) впоследствии показали, что дети с гидантоиновым синдромом действительно имеют более низкую активность эпоксидгидролазы.Эпоксидгидролаза могла бы служить для детоксикации промежуточного соединения оксида арена фенитоина, и было высказано предположение, что пониженная активность этого фермента ответственна за повышенную чувствительность к фенитоину. Hassett et al. (1994) сообщили о полиморфизме гена эпоксидгидролазы, который заметно снижает активность фермента. И наоборот, исходное соединение DPH может быть тератогенным, и генетические дефекты в CYP2C9 и CYP2C19 (двух ферментах, метаболизирующих DPH) могут привести к накоплению тератогена DPH.Частота полиморфизмов с плохим метаболизмом CYP2C9 и CYP2C19 колеблется от 10% до 25% людей в разных популяциях, что очень похоже на процент женщин, принимающих DHP, у которых есть дети с синдромом гидантоина плода.
Альдегиддегидрогеназа 2, алкогольдегидрогеназа 2 и предрасположенность к алкогольному синдрому плода: при метаболизме этилового спирта алкогольдегидрогеназа (ADH) катализирует превращение алкоголя в ацетальдегид, а ацетилдегиддегидрогеназа (ALDH) окисляет это превращение. продукт в уксусную кислоту.Считается, что алкоголь и ацетальдегид, но не уксусная кислота, могут иметь пагубные последствия. Люди обладают как минимум семью генами ADH и 13 генами ALDH. Крабб (1990) указал, что мутация единственного основания в ALDh3 (митохондриальная, в отличие от цитозольной ALDH), которая отвечает за острую реакцию прилива алкоголя и непереносимость алкоголя в основном у азиатов, является наиболее хорошо охарактеризованным генетическим фактором, влияющим на употребление алкоголя. поведение (снижение активности, коррелирующее с непереносимостью).Он высказал предположение, что полиморфизм в нескольких генах алкогольдегидрогеназы может быть связан с риском алкогольного синдрома плода (FAS). Генетическое влияние алкогольного синдрома плода подтверждается исследованиями на близнецах: Streissguth и Dehaene (1993) установили, что степень соответствия для диагностики алкогольного синдрома плода составляет 5 из 5 для монозиготных и 7 из 11 для дизиготных близнецов. В двух парах с дизиготом у одного близнеца был ФАС, а у другого — эффекты алкоголя на плод (ФАЭ). В двух других дизиготных парах у одного близнеца не было явных отклонений от нормы, а у другого — FAE.Показатели коэффициента интеллекта были наиболее сходными в парах монозиготных близнецов и наименее сходными в парах дизиготных близнецов, несовместимых с диагнозом. Джонсон и др. (1996) задокументировали аномалии ФАС центральной нервной системы (ЦНС) с помощью магнитно-резонансной томографии. ЦНС и черепно-лицевые аномалии были преимущественно симметричными и центральными или срединными. Авторы заявили, что ассоциация подчеркнула концепцию средней линии как особого поля развития. ЦНС уязвима для неблагоприятных факторов во время эмбриогенеза, роста и развития плода.
Как показывают эти четыре примера, дальнейшие исследования взаимодействий генов с окружающей средой с использованием инструментов молекулярной эпидемиологии, вероятно, дадут важную новую информацию о многофакторных причинах дефектов развития. Два из приведенных выше примеров касаются полиморфизма генов, кодирующих ферменты, участвующие в метаболизме агента, а именно фенитоина или алкоголя, а два примера касаются полиморфизма генов, кодирующих белковые интермедиаты путей передачи сигналов и генетических регуляторных цепей ( TGF α или MSX1 ), которые являются компонентами процессов разработки.
Изучение взаимодействия генов с окружающей средой особенно продвинуто при болезнях, связанных с ДМО, и эта область исследований называется экогенетикой или фармакогенетикой. Есть ферменты, метаболизирующие фазу I и фазу II. Ферменты фазы I катализируют превращение экзогенного агента в модифицированную форму, часто в окисленную форму. В некоторых случаях экзогенный агент токсичен, а промежуточный продукт нет, но в других случаях агент нетоксичен, а промежуточный продукт токсичен, что является примером метаболического потенцирования или активации.Вероятно, существует несколько сотен видов ферментов фазы I (и кодирующих их генов) у млекопитающих (включая человека). Большинство из них являются членами большого семейства монооксигеназ цитохрома Р450. Считается, что три или четыре типа ферментов P450 метаболизируют 70-80% рецептурных лекарств, принимаемых пациентами, а дефекты ферментов фазы I коррелируют с чувствительностью к лекарствам и опасными побочными эффектами. Ферменты фазы II впоследствии катализируют конъюгацию модифицированного промежуточного продукта с эндогенным безвредным метаболитом, таким как сахар или аминокислота, и конъюгированная форма, которая обычно нетоксична, затем выводится из организма.В нескольких хорошо проанализированных случаях было обнаружено, что пациенты с высокими уровнями фермента фазы I (следовательно, производящими большое количество токсичного промежуточного продукта) и низким уровнем фермента фазы II (следовательно, неспособными избавиться от этого промежуточного продукта) были особенно на риск от химического воздействия. Таким образом, человеческие варианты с измененными уровнями ферментов одной или другой группы или обоих могут иметь ненормальные лекарственные реакции, вплоть до 20- или 30-кратного повышения чувствительности к лекарствам.
В настоящее время известно не менее 60 экогенетических или фармакогенетических различий; многие из них перечислены в.В этом исследовании эпидемиологические методы и геномные методы дополняют друг друга, и прогресс в ближайшем будущем кажется очевидным. Кажется вероятным, что плод находится в группе повышенного риска пороков развития, потому что либо мать, либо плод не могут усваивать химические вещества так же хорошо, как другие, или потому, что они усваивают их лучше.
ТАБЛИЦА 5-1
Классификация неполного списка фармакогенетических или экогенетических различий человека.
Другая большая область исследования корреляции полиморфизмов с дефектами развития — это компоненты самих процессов развития, а именно ключевые компоненты процессов развития, такие как пути передачи сигналов и генетические регуляторные цепи.Эти компоненты являются мишенями для экзогенных агентов, которые ускользают от детоксикации или усиливаются ферментами фазы I. Были прояснены два примера TGF α с курением и MSX1 с дефектами конечностей. В настоящее время, однако, есть несколько хороших примеров, возможно, просто потому, что до недавнего времени информация о процессах развития была недоступна. Поскольку компоненты развития сохраняются во всех типах и хорошо описаны в Drosophila , C.elegans , а теперь и мыши, доступны средства для получения родственных человеческих последовательностей и поиска полиморфизмов. Это исследование будет дополнительно обсуждаться в главах 8 и 9. Фенотипы нулевых мутантов мышей, генерируемые технологией нокаута эмбриональных стволовых клеток (ES), уже внесли существенный вклад в наши знания о том, как изменения в этих генах и путях влияют на развитие. Такого рода исследования быстро развиваются. Полная делеция некоторых компонентов множества путей, лежащих в основе развития, приводит к летальности эмбриона, но в других случаях, когда для компонента существует избыточность генов, удаление компонента приводит к рождению мышей с дефектами развития, напоминающими человеческие дефекты (см. примеры в главе 6).Работа над ключевыми компонентами развития животных может принести большую пользу поиску человеческих вариантов компонентов развития.
Последний шаг, однако, будет заключаться в оценке того, как конкретные токсиканты взаимодействуют с этими измененными путями, вызывая аномальное развитие. Соответствующие гены восприимчивости развития затем могут быть исследованы в человеческих популяциях и могут быть идентифицированы взаимодействия между аллелями этих генов и воздействием токсичных веществ. Будут ли аллельные варианты генов, контролирующих развитие, такие как те, которые кодируют компоненты основных путей передачи сигнала, более важны, столь же важны или менее важны, чем те, которые контролируют DME, еще предстоит определить, и им следует уделить первоочередное внимание в будущих исследованиях.
РЕЗЮМЕ
Секвенирование генома человека и различных геномов животных предоставит фундаментальную информацию об организации генома, эволюции генома, разнообразии последовательностей генов и генетических полиморфизмах. Секвенирование также предоставит платформу для глобального систематического анализа функции генов и экспрессии генов. Ожидается, что токсикология развития и оценка риска в значительной степени выиграют от новых методологий и информации, а именно, при анализе взаимодействий генов и окружающей среды при дефектах развития человека и при анализе воздействия токсичных веществ на процессы развития.
Считается, что от четверти до половины дефектов развития человека связаны с взаимодействием генотипа и окружающей среды, то есть с воздействием на людей определенного генетического состава определенных условий окружающей среды, к которым они более чувствительны, чем другие. . Сложные взаимодействия генов с окружающей средой представляют собой серьезную проблему для токсикологии развития. Для достижения прогресса в понимании взаимодействия генов с окружающей средой потребуются лучшие эпидемиологические методы, наиболее точные молекулярные оценки воздействия и воздействия, а также наиболее подробный анализ генетических различий.Недавние улучшения в высокопроизводительном секвенировании генома человека и в идентификации полиморфных маркеров, удобно расположенных вдоль каждой хромосомы, увеличивают шансы на прогресс в этом направлении. В рамках этой новой области молекулярной эпидемиологии недавние исследования человеческих различий в уровнях активности различных DME и генетической основы этих различий открывают большие перспективы. Другие виды генных продуктов, которые могут иметь важное значение для восприимчивости, но менее известны, включают компоненты процессов развития, особенно компоненты путей передачи сигналов и генетических регуляторных цепей.Эти компоненты будут обсуждаться в следующих главах.
В настоящее время доступны методы для описания моделей одновременной экспрессии тысяч генов развивающихся клеток и тканей и, в принципе, для описания изменений экспрессии в эмбрионах нормальных экспериментальных животных и эмбрионов после тестирования с токсичными веществами. Ожидается, что использование таких методов улучшит категоризацию и анализ дефектов развития, вызванных токсичными веществами.
Объем данных, полученных с помощью современных геномных методов, огромен.Для получения полной выгоды от данных, факультеты или подразделения биоинформатики в университетах и отраслях будут необходимы для отслеживания данных и анализа их в отношении вопросов об организации и функции генома.
Деконструкция источников ассоциаций генотип-фенотип у людей
Abstract
В последние десятилетия попытки связать вариации в геноме человека с фенотипами достигли огромных успехов. Было показано, что на большинство человеческих черт влияет большое количество генетических вариантов в геноме.Для интерпретации этих ассоциаций и их надежного использования — в частности, для фенотипического предсказания — необходимо лучшее понимание многих источников ассоциаций генотип-фенотип. Мы резюмируем прогресс, достигнутый в этом направлении у людей, особенно в разложении прямых и косвенных генетических эффектов, а также в искажении структуры популяции. Мы обсуждаем естественные следующие шаги в сборе данных и разработке методологии, уделяя особое внимание тому, что можно получить, анализируя данные генотипа и фенотипа от близких родственников.
Не так давно генетический анализ проводился с использованием значений признаков (фенотипов) в семьях, без генетических данных. Открытие легко измеряемых геномных маркеров позволило идентифицировать гены болезни с помощью анализа сцепления без предварительного знания основных механизмов ( 1 ). Это привело к идентификации гена, ответственного за Х-сцепленное фагоцитарное расстройство хронической гранулематозной болезни в 1986 году, за которым последовали гены других менделевских заболеваний, таких как муковисцидоз ( 2 ) и болезнь Хантингтона ( 3 ), а также гены рака груди ( 4 , 5 ).Этот подход также применялся для изучения общих сложных заболеваний, включая диабет 2 типа, но не смог предоставить воспроизводимых результатов.
Вторым крупным достижением стало создание массивов однонуклеотидного полиморфизма (SNP) с высокой пропускной способностью, которые позволили одновременно генотипировать сотни тысяч SNP, что привело к исследованию общегеномных ассоциаций (GWAS) ( 6 ) . GWAS проверяет каждый SNP на связь с фенотипом без данных о семье. Успех GWAS начался с открытия, что CFH способствует возрастной дегенерации желтого пятна; этот анализ был основан на 96 случаях и 50 контрольных ( 7 ).Последующее увеличение размера выборки, которая сейчас составляет более 2 миллионов ( 8 ), привело к открытию тысяч генетических вариантов, влияющих на сотни человеческих черт. Результаты GWAS обещают выявить новые лекарственные цели ( 9 , 10 ), среди других приложений.
Способность GWAS идентифицировать SNP, влияющий на признак, зависит от доли вариации признака, объясняемой SNP, которая увеличивается пропорционально квадрату размера эффекта и гетерозиготности.Поскольку гетерозиготность выше для более распространенных вариантов, первоначальные успехи были в основном для вариантов восприимчивости с частотой минорных аллелей выше 5%. Даже если общий вариант не анализируется напрямую, он, вероятно, сильно коррелирует с генотипированным рядом SNP из-за отсутствия событий наследственной рекомбинации между ними. Эта корреляция называется «локальным» неравновесием по сцеплению (LD). Нелокальная LD — корреляция между вариантами, которые не являются физически близкими — может быть результатом неслучайного спаривания.В результате локальной LD GWAS обычно не идентифицирует непосредственно конкретный причинный вариант, а только определяет его приблизительное геномное положение. Для выявления причинных вариантов необходимо мелкомасштабное картирование, которое часто требует функционального анализа и экспериментов ( 11 ).
«Для многих сложных признаков GWAS изменил ландшафт генетических исследований и наше понимание генетической архитектуры»
Большинство общих вариантов, обнаруженных GWAS, влияющих на риск заболевания, имеют эффекты от низких до умеренных (увеличивая вероятность заболевания менее чем в 1 раз.5 на аллель риска) ( 12 , 13 ). Применение GWAS для секвенирования всего экзома и всего генома, наряду со статистическим вменением вариантов на уровне последовательности в образцы, генотипированные с помощью массивов SNP, привело к открытию некоторых более редких вариантов с большим эффектом ( 14 ). Хотя дисперсия признаков, объясняемая общегеномными значимыми (GWS) локусами, увеличилась, для большинства сложных признаков дисперсия, объясняемая локусами GWS, составляет лишь часть предполагаемой наследуемости.Этот пробел, названный «отсутствующей наследуемостью», обсуждается ниже ( 12 , 15 ).
Для сложных признаков идентификация всех причинных вариантов и выяснение лежащих в их основе механизмов остается далекой целью. Однако данные GWAS можно использовать для прогнозирования генотипов, особенно с помощью полигенных оценок (PGS). PGS объединяет предполагаемые эффекты нескольких генетических вариантов, чтобы предоставить прогнозируемое значение признака для человека. Были исследованы многие применения PGS, такие как идентификация людей с существенно повышенным генетическим риском сердечных заболеваний ( 16 ).Несмотря на продемонстрированную ценность PGS, начали появляться вопросы относительно надежности и интерпретации (то есть, что движет предсказательной силой) ( 17 , 18 ).
В GWAS широко признано, что ассоциации могут быть искажены стратификацией населения, в первую очередь ассоциацией между происхождением и влиянием окружающей среды. Методы с поправкой на происхождение вместе с репликацией ( 19 ) дают уверенность в том, что большинство ассоциаций GWS с общими SNP являются истинно положительными.Однако это не означает, что систематическая ошибка устранена, а природа ассоциаций генотип-фенотип должным образом охарактеризована. Мы стремимся изложить здесь различные вклады в ассоциацию генотип-фенотип, объяснить трудности, которые они создают, и предложить возможные решения.
Эффекты, фиксируемые ассоциациями GWAS
Связь между генетическим вариантом и фенотипом может быть разложена на прямое влияние варианта, косвенное генетическое влияние варианта и смешанные эффекты (рис.1). Примером может служить вариант, который оказывает прямое влияние на уровень образования (ОА) при наследовании, а также косвенный эффект через поведение / воспитание родителей ( 20 ). Тот же вариант может иметь косвенное влияние на здоровье через родительское воспитание, но практически не влияет. Прямые эффекты включают широкий спектр причинно-следственных связей, некоторые из которых непростые и не «прямые»; например, варианты в CHRNA5 влияют на риск рака легких через их связь с количеством выкуриваемых ( 21 ).Кроме того, прямой эффект здесь может включать эффекты других вариантов локальной LD. Обратите внимание, что типичный GWAS, проводимый без данных о семье, позволяет оценить только сумму прямого и косвенного воздействия (комбинированный эффект), а не их по отдельности.
Рис. 1 Сигналы, захваченные GWAS отдаленно связанных людей и семей.На основе удаленных родственников оценки величины воздействия SNP на признак включают прямые генетические эффекты (черный цвет), а также ряд других эффектов, в том числе искажение из-за структуры популяции (серый цвет), выборочное спаривание по признаку или коррелированный (бордовый), косвенные генетические эффекты от родителей (синий) и сиб-эффекты (персик).GWAS на основе семьи (например, использование трио) использует родительские генотипы в качестве контроля для отделения прямых от косвенных генетических эффектов и других смешивающих эффектов ( 20 ), как показано в разложении справа. На этом рисунке мы игнорируем эффекты локального LD.
КРЕДИТ: KELLIE HOLOSKI / SCIENCE
В рамках аддитивной модели для совместных эффектов вариантов мы определяем генетический компонент как линейную комбинацию генотипов всех причинных вариантов с весами, пропорциональными истинным (прямым, косвенным , или комбинированные) эффекты (рис.2). Генетические компоненты прямого и косвенного воздействия различны, но они могут коррелировать с силой, которая зависит от генетической корреляции между интересующим фенотипом пробанда и фенотипами родственников, через которые опосредуются косвенные эффекты. Например, эта корреляция, вероятно, сильна для EA и слабая для индекса массы тела (ИМТ) ( 20 ). Относительные силы этих двух генетических компонентов и их корреляция определяют корреляции с генетическим компонентом комбинированного эффекта.Поскольку PGS, построенный на основе типичного GWAS, использует оценки комбинированных эффектов, его предсказательная сила иногда может быть значительно сильнее, чем то, что можно объяснить только прямыми эффектами ( 20 ).
Рис. 2 Генетические модели с двумя признаками, объединенные или разделенные прямым и косвенным эффектами.Для признака, предполагая аддитивную модель, генетический компонент, объединяющий прямые и косвенные эффекты, равен g δ + η = ∑ i (δ i + η i ) g i , где g i , δ i и η i обозначают генотип, прямое и косвенное влияние варианта i соответственно.Вверху: с двумя характеристиками (1 и 2) есть две величины и одна корреляция. Для каждого признака комбинированный генетический компонент может быть разделен на компонент прямого действия, g δ = ∑ i δ i g i , и компонент косвенного воздействия, g η = ∑ i η i g i . Внизу: модель с двумя признаками становится одной с четырьмя генетическими компонентами и шестью попарными корреляциями между ними.Для проиллюстрированного здесь канонического примера, где признак 1 может быть EA, а признак 2 может быть BMI, размер точки указывает величину компонента, а толщина соединительной линии указывает силу корреляции.
Генетические эффекты могут способствовать ассоциации между чертами через плейотропию. Модель с двумя признаками плейотропии (рис. 2, вверху) комбинированных эффектов имеет три параметра: дисперсии, объясняемые генетическими компонентами комбинированного эффекта двух признаков, и корреляцию между ними.Эта корреляция была оценена для многих пар признаков с использованием данных GWAS ( 22 ). Разделив компоненты прямого и косвенного воздействия, модель (рис. 2, внизу) имеет 10 параметров, включая величины четырех генетических компонентов прямого и косвенного воздействия и шесть корреляций. Полная модель не может быть оценена с помощью стандартного GWAS, поэтому в настоящее время у нас мало понимания того, в какой степени прямые и косвенные генетические эффекты по-разному влияют на плейотропию.
Смешивающие эффекты
Связь между генетическим вариантом и фенотипом может частично отражать корреляцию с каким-либо другим причинным феноменом (средовым или генетическим), а не истинным причинным эффектом SNP на фенотип.Этот тип смешения возникает из-за наличия неслучайного спаривания, приводящего к структуре населения. В GWAS есть по крайней мере три источника искажения: (i) искажение окружающей среды, когда частоты аллелей и эффекты среды коррелированно меняются в разных географических регионах или субпопуляциях; (ii) генетическое смешение, когда различия частот аллелей между субпопуляциями коррелируют с частотными различиями других аллелей с причинными эффектами; или (iii) смешивание ассортативного спаривания, которое происходит, когда есть ассортативное спаривание по признаку (или коррелированному признаку), вариант с причинным эффектом на признак становится коррелированным с другими вариантами с причинными эффектами, и его связь с признаком затем фиксирует свой собственный причинный эффект плюс часть других вариантов.Эти формы смешения концептуально различны, но на практике они часто взаимосвязаны.
Поправка на искажение в GWAS
Корректировка главного компонента (PC) — это распространенный метод, используемый для устранения некоторых искажающих эффектов, связанных со структурой популяции ( 23 ). В идеале основные компоненты, используемые для корректировок, сильно коррелируют с компонентом, влияющим на окружающую среду, и не коррелируют с компонентом прямого генетического воздействия. Если компонент прямого воздействия существенно коррелирует с искажающими компонентами, корректировка ПК удалит некоторые прямые генетические эффекты, а также смешанные эффекты.
Смешивающий компонент ассортативного спаривания (iii) по своей природе почти идеально коррелирует с суммой прямых и косвенных компонентов. Ассортативное спаривание по таким признакам, как рост и EA ( 24 ) приводит к нелокальному LD вариантов с прямыми и косвенными эффектами, которые улавливаются PC. Таким образом, теоретически настройка ПК может отрегулировать большую часть компонента прямого воздействия. На практике этого не происходит. Даже при очень большом размере выборки предполагаемые ПК, скорее всего, будут в основном шумными, помимо нескольких сильных (часто географических) сигналов.Результаты выборки белых британцев (WB) UKB подчеркивают этот момент (рис.3): помимо первых восьми самых сильных ПК, ПК, вычисленные на основе выборки из 272 519 человек ( 25 ), по-видимому, в основном обусловлены шумом выборки и локальным LD. в хромосомах. Шум может маскировать тонкую структуру населения, что может привести к искажению GWAS даже после настройки ПК ( 26 ).
Рис. 3 Поведение основных компонентов 272 519 образцов биобанка Великобритании.Мы исследуем степень, в которой основные компоненты отражают реальную структуру популяции, исследуя, воспроизводится ли генетическая дисперсия (собственные значения), объясняемая 40 основными компонентами, выведенными из 146082 SNP в 272 519 британских образцах биобанка белых британцев (WB), в независимой выборке ВБ.Собственное значение репликации выше 1 указывает на то, что предполагаемый главный компонент фиксирует воспроизводимые корреляции между SNP, либо локальной LD (в пределах хромосомы), либо структурой популяции (в основном между хромосомами). Исходный (черные кружки): собственные значения главных компонентов в исходном наборе из 272 519 человек из ВБ. Репликация (синие треугольники): эквиваленты собственных значений, то есть дисперсии линейных комбинаций генотипов SNP с использованием весов, выведенных из исходного набора, и стандартизованных генотипов в наборе репликации из 64 969 индивидуумов WB.Репликация (только между хромосомами) (красные кресты): с использованием того же набора репликации, но эквиваленты собственных значений, вычисленные путем игнорирования ковариаций пар SNP в одних и тех же хромосомах и подсчета только ковариаций пар SNP на разных хромосомах, что включает 94,8% все пары SNP. Среднее собственное значение для последних 32 ПК снижается с 4,37 для исходного набора до 2,61 для набора репликации и далее до 1,03 для набора между хромосомами, указывая на то, что эти ПК в основном улавливают шум и локальную LD, а не структуру популяции.
Фитинг линейных смешанных моделей (LMM) является альтернативой настройке с помощью ПК. Эти методы выполняют тип регрессии для набора SNP, где эффект каждого SNP моделируется как «случайный эффект», полученный из нормального распределения ( 27 ). LMM уже давно используются для прогнозирования признаков в селекции животных ( 28 ). В исследованиях на людях тестирование ассоциации LMM обычно состоит из оценки эффекта фокусного SNP как «фиксированного эффекта» при моделировании случайных эффектов для набора других SNP.Наивные вычисления LMM масштабируются с кубом размера выборки, и поэтому были разработаны альтернативные вычислительные подходы для обработки больших наборов выборок GWAS ( 29 ).
Привлекательность LLM заключается в том, что они позволяют улучшить моделирование стратификации населения и родства выборки ( 27 ). LMM часто используются в сочетании с корректировкой PCA и могут учитывать более сложные модели стратификации путем моделирования эффектов (почти) всех измеренных SNP, фиксируя как реальные генетические эффекты, так и эффекты стратификации ( 27 ).Кроме того, методы LMM могут привести к улучшенной оценке эффектов SNP и их ошибок выборки по сравнению с линейной регрессией при наличии родства выборки ( 27 ). LMM могут также уменьшить смещение в оценках эффекта SNP из-за ассортативного спаривания ( 30 ). Однако современные методы LMM GWAS не устраняют косвенных генетических эффектов.
Использование данных генотипа семьи
Учитывая родительские генотипы, генотип потомства определяется путем случайной сегрегации генетического материала во время мейоза.Эта случайная сегрегация не коррелирует с косвенными генетическими эффектами от родственников и другими смешивающими эффектами. Таким образом, родительские генотипы можно использовать в качестве контроля для получения объективных оценок прямых генетических эффектов ( 20 , 31 ) (рис. 1). Точно так же генетические различия между братьями и сестрами являются результатом случайной менделевской сегрегации родителей во время мейоза. Следовательно, генетические различия между братьями и сестрами не смешиваются с косвенными генетическими эффектами родителей, стратификацией популяции и ассортативным спариванием.Однако методы, использующие различия в генотипах братьев и сестер, оценивают прямой эффект за вычетом косвенного эффекта от брата или сестры, и, следовательно, дают объективные оценки прямого эффекта только тогда, когда косвенный генетический эффект родства равен нулю.
Изучение косвенных генетических эффектов имеет долгую историю в животноводстве ( 13 ). У людей в большинстве исследований косвенных генетических эффектов использовались PGS, полученные из GWAS, которые не различают прямые и косвенные генетические эффекты (рис.4) ( 20 ). Однако, когда прямые и косвенные генетические эффекты не полностью коррелируют, этот подход не может дать полной картины ( 32 ). В идеале GWAS следует проводить с генотипами родителей или братьев и сестер в качестве контроля и с использованием моделей с косвенными генетическими эффектами. Однако возможности этого подхода в настоящее время ограничены, поскольку большие выборки с генотипированными братьями и сестрами и / или родителями встречаются редко. Кроме того, поскольку только около половины генетической изменчивости в популяции является внутрисемейной, для получения такой же исследовательской мощности, как и при стандартном GWAS-анализе, требуются существенно большие выборки семей.Следовательно, необходимы методы, объединяющие информацию из стандартных GWAS и из анализа семейств.
Рис. 4 Сокращение полигенного прогноза и оценок наследуемости с использованием внутрисемейных планов с использованием исландских данных.( A ) Оценка наследуемости SNP с использованием переданных аллелей дается hSNP2; оценка наследуемости SNP с использованием внутрисемейного метода, регрессии неравновесия по родству (RDR) ( 31 ), дается с помощью hRDR-SNP2.Статистически значимые различия ( P <0,05, односторонний z-тест) наблюдались для EA hSNP2 / hRDR-SNP2 = 1,72 ( P = 7,6 × 10 −3 ) и роста hSNP2 / hRDR-SNP2 = 1,24 ( P = 0,015). ( B ) Дисперсия, объясняемая регрессией признака на полигенную оценку, дается Rpoly2; дисперсия, объясняемая полигенной оценкой, когда ее эффект оценивается с использованием внутрисемейного (трио) дизайна, определяется как Rpoly: δ2 ( 20 ). Мы подчеркиваем относительный размер оценок от внутрисемейных методов (hRDR-SNP2 и Rpoly: δ2) до межсемейных методов (hSNP2 и Rpoly2).Межсемейные методы фиксируют косвенные генетические эффекты от родственников и, возможно, стратификацию популяции и ассортативное спаривание в дополнение к наследуемости, зафиксированной внутрисемейными методами. Сокращения для признаков: ИМТ, индекс массы тела; EA, уровень образования (лет).
Наследственность
Традиционно наследуемость оценивалась путем сравнения корреляций между однояйцевыми и неидентичными близнецами. Помимо идентификации конкретных причинных локусов, можно использовать данные GWAS для оценки фенотипической изменчивости, объясняемой генетической изменчивостью, зафиксированной с помощью SNP (и вариантов в LD с ними) на массиве генотипирования, называемом «наследуемость SNP» или hSNP2 ( 33 ).Оценки hSNP2 подразумевают, что общие генетические варианты, проанализированные на типичном массиве генотипов, в совокупности объясняют значительно большую фенотипическую дисперсию, чем варианты GWS. Однако оценки hSNP2, как правило, значительно ниже оценок наследуемости из исследований близнецов ( 15 ), что является частью «проблемы отсутствия наследственности». Некоторые, но далеко не все, этот разрыв объясняется эффектами вмененных вариантов, которые не находятся в сильной LD с маркерами на типичном массиве генотипов ( 13 , 34 ).Одна из возможностей состоит в том, что большая часть оставшейся недостающей наследуемости объясняется очень редкими вариантами ( 35 ).
Широко используемый метод, GREML, оценивает hSNP2 путем измерения силы взаимосвязи между фенотипическим сходством и геномным генетическим сходством (оценивается по SNP), которая варьируется даже для отдаленных родственников, обычно используемых в GWAS ( 36 ) . Этот подход обеспечивает оценку общей дисперсии, объясняемой комбинированными прямыми и косвенными эффектами аллелей пробандов ( 20 , 31 ).Степень, в которой косвенные генетические эффекты и стратификация популяции повлияли на оценки hSNP2 (рис. 4), не известна, равно как и систематическая ошибка, вызванная ассортативным скрещиванием при оценке наследуемости как внутри семьи, так и между семьей.
Также важно отметить, что общая дисперсия, объясняемая комбинированными прямыми и косвенными эффектами, отличается от традиционно определенной наследуемости, которая касается только прямых эффектов. Тем не менее, это интересный параметр, поскольку он определяет верхнюю границу генетического предсказания по аллелям пробандов.Подразумевается, что верхний предел генетического предсказания признака часто может быть больше, чем наследуемость ( 18 ).
Некоторые недавние методологические разработки
Регрессия оценки LD
С появлением GWAS были разработаны подходы, позволяющие лучше использовать и интерпретировать их результаты. Примечательно, что регрессия оценки LD (LDSC) была разработана для того, чтобы отличить эффекты смешения из-за стратификации населения от причинно-следственных генетических эффектов на статистику теста GWAS ( 37 ).Предполагая высокополигенную архитектуру, ожидается, что статистика теста GWAS для отдельного SNP будет увеличиваться с его баллом LD (мера генетической вариации, помеченной SNP через локальную LD) из-за увеличения корреляции с причинными вариантами. Однако средняя тестовая статистика по всем SNP повышается за счет стратификации популяции из-за корреляции между аллелями и различий в средних значениях признаков между субпопуляциями ( 37 — 39 ). Оценивая, насколько искажение, вызванное стратификацией населения, увеличивает среднюю статистику теста, точку пересечения LDSC можно использовать для корректировки статистики теста GWAS.LDSC также можно использовать для оценки корреляции между эффектами SNP на разные признаки ( 22 ), для разделения вкладов в наследуемость SNP от разных функциональных категорий вариантов ( 40 ) и для облегчения многопоточного метаанализа ( 41 ). ).
Ключевым предположением LDSC является то, что различия частот аллелей между субпопуляциями не зависят от баллов LD ( 37 ). Однако корреляция между оценками LD и различиями частоты аллелей может быть вызвана формами связанного отбора, такими как фоновый отбор ( 26 ).Таким образом, остаются вопросы о надежности меры смещения стратификации населения LDSC.
Менделевская рандомизация
Менделевская рандомизация (MR) использует генетические данные для улучшения причинно-следственных связей в эпидемиологии ( 42 ). Если генетический вариант влияет на признак A, а признак A влияет на признак B, то ожидается, что варианты, которые влияют на признак A, будут влиять на признак B. причинное влияние признака A на признак B, учитывая, что генетические варианты влияют на признак B только через свое влияние на признак A, и что генетические варианты не коррелируют с какими-либо смешивающими факторами.MR доказал свою эффективность в опровержении ложных причинно-следственных гипотез, основанных на данных наблюдений, таких как связь между уровнем холестерина ЛПВП и сердечно-сосудистыми заболеваниями ( 43 ) и сниженным риском сердечно-сосудистых заболеваний у умеренно пьющих в западных обществах ( 44 ).
MR обычно полагается на оценки эффекта SNP из GWAS без семей, которые могут быть искажены стратификацией популяции, косвенными генетическими эффектами от родственников и ассортативным спариванием ( 45 ).Для решения этих проблем были предложены внутрисемейные методы МРТ, которые показали, что предыдущие оценки МР причинных эффектов роста и ИМТ на ЭА были ложными ( 45 ).
Еще одной проблемой для MR-анализа является широко распространенная плейотропия: если SNP влияет на признак B через признак, отличный от признака A, то он не является действенным инструментом для вывода причинного воздействия признака A на признак B. разработаны для решения этой проблемы, их эффективность может зависеть от предварительных знаний о путях смешивания ( 46 ).
Взаимодействие генов с окружающей средой
Взаимодействие генов с окружающей средой (GxE) происходит, когда влияние генетического варианта на признак различается в разных средах ( 47 ). Такие взаимодействия GxE отличаются от корреляции ген-среда, которая может быть результатом, например, косвенных генетических эффектов от родственников. У людей хорошо реплицированные примеры взаимодействий GxE редко встречаются вне фармакогеномики ( 48 , 49 ). Единственным исключением является взаимодействие между вариантами в локусе FTO и физической активностью, влияющей на ИМТ ( 50 , 51 ).
Возможности обнаружения взаимодействий GxE в исследованиях GWA, вероятно, были низкими из-за небольшого размера эффекта и нагрузки на несколько тестов. Один из способов увеличить мощность обнаружения GxE — это поиск взаимодействий между факторами окружающей среды и PGS ( 52 , 53 ). Этот метод эффективен, когда генетические варианты, влияющие на признак, аналогичным образом взаимодействуют с факторами окружающей среды, но не могут идентифицировать взаимодействия между факторами окружающей среды и конкретными генетическими вариантами.LMM могут применяться для обнаружения компонента фенотипической дисперсии, возникающего из-за взаимодействия между геномными генетическими вариантами и фактором окружающей среды ( 54 ), но не могут точно определить взаимодействия с конкретными генетическими вариантами. Генетические варианты, участвующие во взаимодействиях GxE, влияют на изменчивость признака ( 55 , 56 ), что может быть использовано для сокращения пространства поиска потенциальных взаимодействий путем ограничения вариантов с доказательствами влияния на фенотипическую изменчивость.Однако остаются методологические проблемы: эффекты взаимодействия и генетические эффекты на фенотипическую изменчивость чувствительны к шкале измерения ( 56 , 57 ), а влияние стратификации популяции на оценки GxE недостаточно охарактеризовано. Более того, причинность эффектов взаимодействия GxE установить сложно, поскольку взаимодействие может быть связано с неизмеряемым фактором окружающей среды, который коррелирует с измеренным фактором (факторами) окружающей среды, а более широкие социально-экологические факторы, которые могут структурировать воздействие окружающей среды, часто неизвестны.
Переносимость фенотипического предсказания
Точность предсказания на основе PGS зависит от наследуемости признака и мощности существующего GWAS (особенно от размера выборки и генетической архитектуры) ( 28 ). Для некоторых характеристик [таких как рост, для которого текущая точность прогноза составляет ~ 25% ( 58 )], существующие оценки уже являются информативными для групп людей, аналогичных тем, для которых проводился GWAS.
Полигенные оценки не работают также при прогнозировании фенотипов людей, которые отличаются от тех, которые включены в набор GWAS.Некоторые причины понятны и возникают из-за различий в происхождении. Примечательно, что поскольку PGS состоит из взвешенной суммы количества аллелей и поскольку частоты аллелей варьируются по всему миру (из-за генетического дрейфа и естественного отбора), аллели, которые вносят вклад в вариацию признаков в GWAS, с меньшей вероятностью присутствуют или могут даже отсутствовать. у более дальних родственников. Также ожидается, что точность предсказания PGS будет снижаться по группам предков, потому что GWAS не идентифицирует причинные сайты, а наборы возможных причинных сайтов в локальной LD; поскольку локальные паттерны LD зависят от истории популяции, ассоциации, наблюдаемые в одной популяции, будут иметь тенденцию хуже улавливать причинные SNP в других.Как и ожидалось, недавние исследования сообщают, что прирост R 2 для широкого диапазона признаков ниже у людей, чьи предки отличаются от предков группы GWAS ( 59 , 60 ).
Помимо частоты аллелей и различий LD, другие факторы могут способствовать снижению прогностической способности PGS: степень дисперсии окружающей среды может различаться среди групп разнородных предков или отобранных по различным критериям включения ( 18 ), а измерение фенотипа может отличаться. по группам.Более того, величина эффекта вариантов может различаться в результате взаимодействий ген-ген (GxG) и GxE. Изменения в величине эффекта могут быть особенно важны для признаков, на которые косвенные эффекты или ассортативное спаривание вносят большой вклад, поскольку такие факторы могут зависеть от культурных и экологических факторов. Здесь становится важным разложить природу сигналов, идентифицированных в GWAS, чтобы определить, какие компоненты (например, прямые и косвенные эффекты) обеспечивают более легко обобщенные прогнозы.
Outlook
Для многих сложных признаков GWAS изменил ландшафт генетических исследований и наше понимание генетической архитектуры. Если раньше не было ни одной надежно воспроизводимой ассоциации, теперь существуют тысячи вариантов с надежными ассоциациями. Примечательно, что GWAS не требует данных о семье, что облегчает сбор больших размеров выборки. Однако в последнее время на первый план снова выдвигаются уникальные свойства семейных данных. Во-первых, некоторые редкие варианты с сильными эффектами существуют только в расширенных семьях.Наиболее важно то, что для более глубоких и тонких вопросов могут потребоваться семейные данные, такие как тройки родителей и потомков и пары братьев и сестер, чтобы отличить прямые эффекты от косвенных и других смешивающих факторов. Статистически одно естественное расширение — это расширение исследуемой единицы от индивидуума до нуклеарной семьи. В этой связи стоит отметить, что по мере увеличения размера выборки неизбежно будут собираться близкие родственники, поскольку в выборку входят более крупные фракции населения.
Остающейся проблемой является проблема смещения при установлении, которая возникает, когда исследуемые выборки систематически отличаются от совокупности.Большинство выборок смещено в сторону лиц европейского происхождения ( 60 ), а также в отношении лиц с более высоким социально-экономическим статусом и здоровьем ( 61 ), наряду с другими неизвестными предубеждениями. Хотя эти предубеждения при установлении не обязательно приводят к ложноположительным результатам, они ограничивают переносимость результатов GWAS ( 18 , 60 ). Особенно заметными в этом отношении являются взаимодействия GxE не только в пространстве, то есть между популяциями в данный момент времени, но и во времени, учитывая массивные вековые тенденции в окружающей среде, которые произошли и продолжают происходить.Это соображение относится к чертам здоровья, чертам, связанным с образованием, и чертам плодородия, которые влияют на давление отбора. В связи с этим важно не только собирать образцы разных предков и нынешней среды, но, где это возможно, также собирать данные по нескольким поколениям.
Box 1Глоссарий.
Ассортативное спаривание: Когда пары, производящие потомство, выбирают друг друга на основе определенных фенотипов.
Картирование в мелком масштабе: Относится к подходам, которые направлены на определение того, какой вариант или варианты, вероятно, будут причинными среди набора связанных вариантов, идентифицированных в GWAS.
Наследственность: Измеряет долю фенотипической изменчивости, объясняемую прямым воздействием всех генетических вариантов в популяции в данный момент времени.
Гетерозиготность: Вероятность того, что два аллеля на сайте различаются; в предположении равновесия Харди-Вайнберга и с учетом двуаллельного сайта эта мера генетического разнообразия определяется как 2 p (1 — p ), где p — частота аллеля.
Imputation: Статистический метод, который выводит генотипы людей в вариантах, не измеренных напрямую в массиве генотипов, путем ссылки на полные данные последовательности генома.
Косвенный генетический эффект: Влияние генетического варианта у одного человека на черты другого человека через окружающую среду.
Общегеномные значимые ассоциации (GWS): Варианты, связанные с фенотипом на уровне значимости, выбранном для преодоления бремени множественного тестирования, обычно устанавливается на P <5 × 10 –8 .
Анализ сцепления: Тесты на совместную сегрегацию фенотипов и генотипов внутри семей.
Плейотропия: Распространенное наблюдение, что многие SNP, связанные с одним признаком, также связаны с другими признаками. Относится к концепции генетической корреляции.
Главный компонент: Главный компонент — это предполагаемая ось генетической изменчивости в выборке. Главный компонент — это линейная комбинация генотипов SNP, где каждый SNP имеет «нагрузку», дающую свой вклад в главный компонент.
Полигенная оценка (PGS): Взвешенная сумма аллелей, переносимых индивидуумом, где веса даны по величине эффекта, оцененной в GWAS.
Благодарности: Спасибо Д. Конли, А. Харпаку и Дж. Притчарду за комментарии к черновику рукописи. При поддержке Фонда Ли Ка Шинга (A.I.Y., S.B. и A.K.) и гранта NIH R01 GM121372 (M.P.). Это исследование было проведено с использованием ресурса UK Biobank Resource под номером заявки 11867.
Генетические различия: генотип и фенотип
2.12 По последним лучшим оценкам, все люди имеют одинаковый базовый набор из примерно 32 000–35 000 генов. [15] Это намного ниже первоначальных оценок в 200 000 и даже относительно недавних оценок в 100 000, использованных в начале Проекта генома человека. Эта цифра аналогична для мыши — и, по крайней мере, для некоторых людей, неудобно близка к цифрам для круглого червя (19 000), плодовой мушки (13 000) и горчичного кресс-салата (25 000).Как широко сообщалось, геном человека более чем на 98% идентичен геному шимпанзе и на 97% идентичен геному горилл. [16]
2.13 Гены могут иметь разные версии, известные как аллели . Эти аллели возникают, когда происходит изменение порядка оснований, описанное выше — по сути, «типографская ошибка» в коде, включающая изменение одной буквы, инверсию двух букв, удаление или вставку фразы. («кодон») или повторение фразы.Это изменение в последовательности может не причинить вреда («полиморфизм») или может сделать ген неисправным («мутация») в том, как он направляет (выражает) производство белка.
2.14 Хотя любые два человека будут на 99,9% генетически идентичны, точная последовательность ДНК из примерно 6,2 миллиарда букв (3,1 миллиарда пар оснований) различается в генетическом коде каждого человека. Считается, что оставшиеся 0,1% различия составляют более 10 миллионов обычных однобуквенных генетических вариаций (и большее количество редких вариантов).Скорость изменчивости у людей очень низкая (один однонуклеотидный полиморфизм на 1300 оснований) по сравнению с другими видами, включая других приматов, что позволяет предположить наличие небольшого вида с небольшой «начальной популяцией». [17]
2.15 Эти факты объясняют как поразительное сходство между всеми людьми, которое является результатом нашей общей наследственности, так и многочисленные индивидуальные различия, обнаруживаемые даже внутри нуклеарной семьи.
2.16 Некоторые генетические вариации практически не влияют на здоровье, например цвет волос.Однако некоторые мутации влияют на базовое функционирование:
Мутации — это постоянные и наследуемые изменения способности гена кодировать свой белок. Подобно типографским ошибкам, которые могут изменить значение слова или даже превратить предложение в тарабарщину, такие изменения в структуре гена могут серьезно повлиять на способность гена кодировать свой белок. Некоторые мутации предотвращают образование любого белка, некоторые производят нефункциональный или только частично функциональный белок, а некоторые производят дефектную или ядовитую версию белка. [18]
2.17 Например, болезнь Хантингтона (HD) вызывается мутацией гена, расположенного на хромосоме 4, в которой триплет «CAG» повторяется аномально большое количество раз.